UREA CYCLE
Normally the adult human is in nitrogen balance. The amount of nitrogen ingested each day, mainly in the form of dietary protein, is equal to the amount of nitrogen excreted. The major nitrogenous excretory product is urea, which is produced in the liver, and exits the body in the urine. Ammonia, produced from the α-amino group of amino acids and other nitrogen-containing compounds in extra-hepatic tissues is toxic, particularly to neural tissue, and must, therefore, be transported in non-toxic form in to the liver for conversion to urea, a non-toxic compound that is excreted by the kidney.Alanine and glutamine are the major transporters of nitrogen in the blood. Alanine is produced in a single biochemical step by the transamination of pyruvate. Glutamine is produced from glutamate by the addition of an amide to the glutamate γ carboxyl group by an ATP-dependent reaction catalyzed by glutamine synthetase.
NH4+ and aspartate, the forms in which nitrogen enters the urea cycle, are produced from amino acids in the liver by a series of transamination and deamination reactions. Glutamate dehydrogenase is a key enzyme in the process because it generates the free NH4+ previously transferred to α-ketoglutarate from many amino acids by transaminases. As dietary protein increases (a protein-rich diet) the concentration of the enzymes of the urea cycle increase, suggesting a regulated response to meet the increased need for nitrogen disposal.
Reactions of the Urea Cycle
Two nitrogen atoms enter the urea cycle as NH4+ and aspartate. The first steps of the cycle take place in liver mitochondria, where NH4+ combines with HCO3- to form carbamoyl phosphate. Carbamoyl phosphate reacts with ornithine, a compound both required as input to, and regenerated by the cycle, to produce citrulline, which, exits the mitochondria to the cytosol, where the remaining reactions of the cycle occur. The amino acid arginine is synthesized as a product of the urea cycle. Fumarate, another product, links the urea cycle with the TCA cycle. The two entering nitrogen atoms exit the cycle as urea, which the liver releases into the blood for disposal, in urine, by the kidneys.
- Synthesis of carbamoyl phosphate by Carbamoyl Phosphate Synthetase I
- in mitochondria of the liver
- one of only three reactions in humans that can “fix” NH4+, i.e., covalently link it to carbon
- NH4+, CO2 (as bicarbonate) and 2 ATP react to form carbamoyl phosphate.
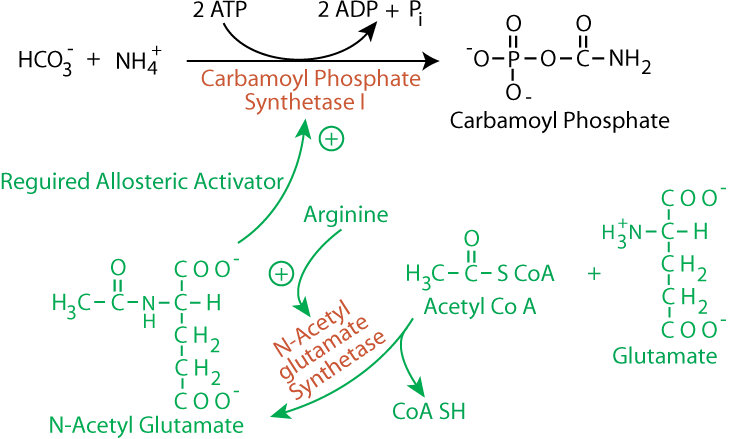 2 ATP molecules provide the energy to create the phosphoanhydride and N-C bonds of carbamoyl phosphate; inorganic phosphate and 2 ADP produced.
2 ATP molecules provide the energy to create the phosphoanhydride and N-C bonds of carbamoyl phosphate; inorganic phosphate and 2 ADP produced.- stimulated by N-acetyl-glutamate (a required allosteric activator), which is synthesized from acetyl CoA and glutamate; the synthesis of N-acetyl-glutamate is stimulated by arginine, the immediate precursor of urea in the urea cycle. Increased levels of amino acids, signaled by increased arginine levels, therefore, stimulate urea production by the urea cycle.
- NOTE: Carbamoyl phosphate synthetase I is present in liver mitochondria and uses NH4+ as a source of nitrogen; carbamoyl phosphate synthetase II is present in the cytosol of many cells, uses glutamine as a source of nitrogen, and produces carbamoyl phosphate for pyrimidine biosynthesis.
- Synthesis of citrulline from carbamoyl phosphate and ornithine by Ornithine Transcarbamoylase
- X-linked gene
- in mitochondria; ornithine transported into mitochondria
- carbamoyl phosphate is the carbamoyl donor which has a high transfer potential because of its phosphoanhydride bond
- inorganic phosphate released
- citrulline produced, which is transported from the mitochondria to the cytosol where the remaining reactions of the urea cycle occur
- Synthesis of argininosuccinate by condensation of citrulline and aspartate by Argininosuccinate Synthetase
- driven by the cleavage of ATP; AMP and inorganic pyrophosphate produced; inorganic pyrophosphate cleaved by cellular pyrophosphatases to inorganic phosphate
- Argininosuccinate cleaved by Argininosuccinase to produce fumarate and arginine
- NOTE: The carbon skeleton of aspartate is conserved as fumarate, with transfer of the aspartate amino group to arginine. Recall that fumarate is a TCA cycle intermediate, and can be hydrated to form malate. In the fed state malate may be converted by malic enzyme to pyruvate, which serves as a source for the synthesis of fatty acids. It may also be oxidized to oxaloacetate. Oxaloacetate can have several fates. It can be transaminated to aspartate (aspartate transaminase), combine with acetyl CoA to enter the TCA cycle or, in the starved state, be converted to phosphoenolpyruvate for gluconeogenesis.
- Urea production and the regeneration of ornithine from arginine by Arginase
- urea passes into the blood and is eliminated by the kidneys,
- urea accounts for approx. 90% of all bodily nitrogenous excretory products.
- ornithine is synthesized from glucose; arginine is synthesized from ornithine by the urea cycle
Click The Image
Nitrogen, As An Ammonium Ion, Is Fixed To The Bicarbonate Carbon
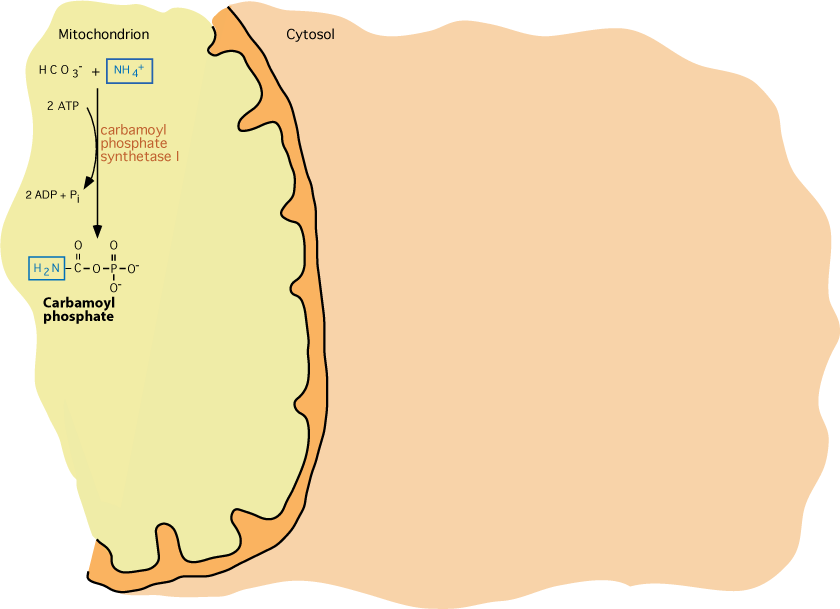
Carbamoyl Phosphate Synthetase I — The regulated step of urea synthesis, occurs in mitochondria, where 2 molecules of ATP are used to "fix" nitrogen to the carbon donated by the bicarbonate ion. The product, carbamoyl phosphate, is the same as that produced by Carbamoyl Phosphate Synthetase II, a cytoplasmic enzyme in the pyrimidine synthetic pathway, which uses glutamine as the nitrogen donor instead of the ammonium ion.
X-Linked Ornithine Transcarbamoylase
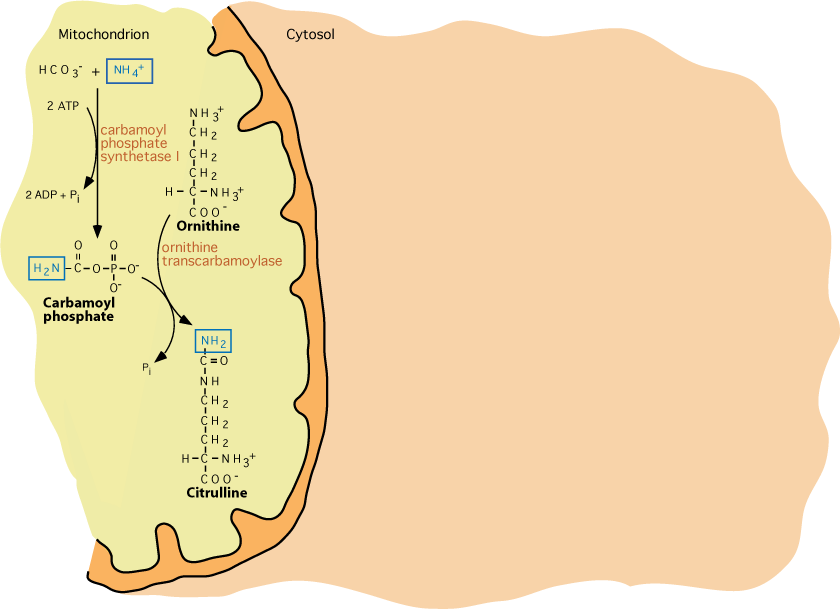
Ornithine, synthesized from glutamate, reacts with carbamoyl phosphate in a reaction catalyzed by Ornithine Transcarbamoylase, whose gene resides on the X chromosome. Of all defects in urea cycle enzymes, defects of ornithine transcarbamoylase are the most frequent.
Citrulline Exits The Mitochondria And Acquires A Second Nitrogen
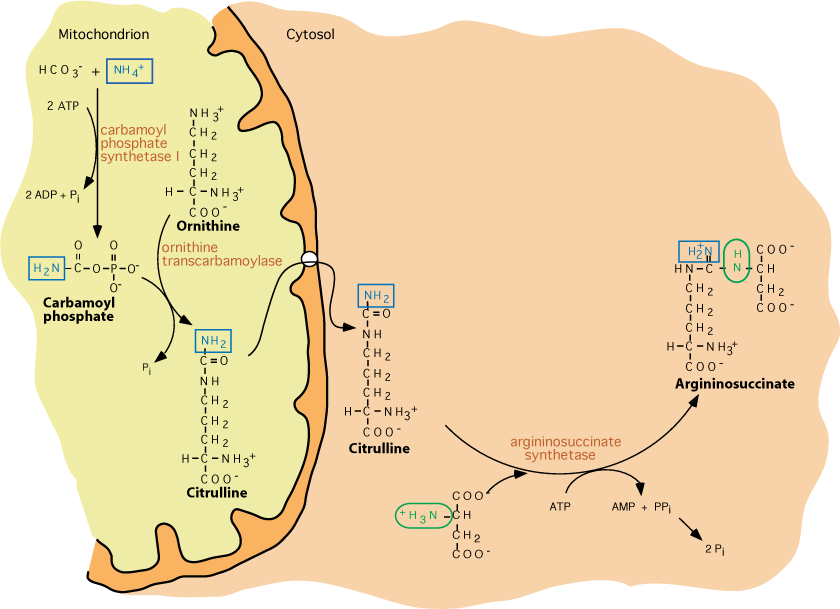
Citrulline is transported out of the mitochondrion to the cytosol, where it reacts with aspartate to yield argininosuccinate in a reaction catalyzed by Argininosuccinate Synthetase, which requires energy from the hydrolysis of ATP to AMP and Pi.
Arginine Is Synthesized
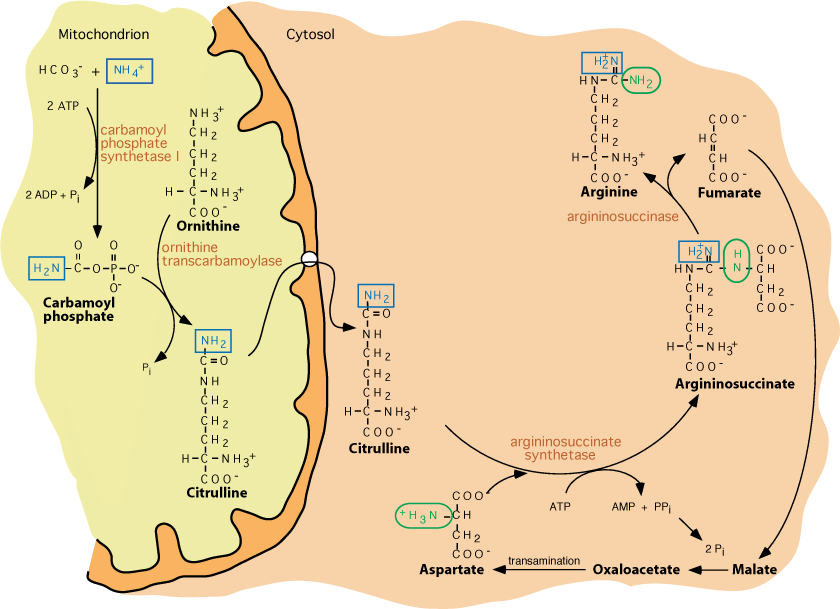
Argininosuccinase releases the aspartate carbon skeleton as fumarate, a TCA cycle intermediate, but not the aspartate nitrogen, to yield the amino acid arginine. Fumarate can be converted to oxaloacetate, another TCA cycle intermediate, which can be transaminated to another molecule of aspartate that can react with another molecule of citrulline and carry another nitrogen into the urea cycle.
Urea Is Formed And Ornithine Is Regenerated
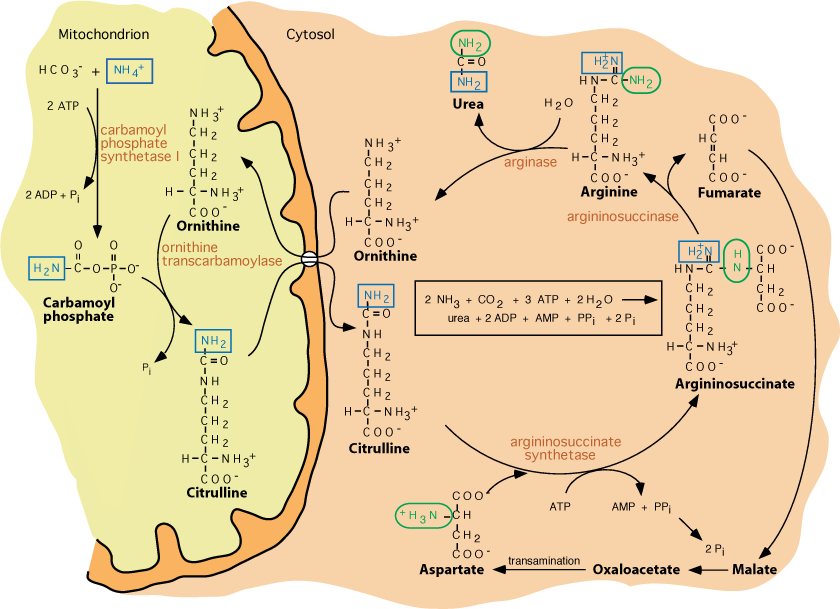
Arginase hydrolyzes arginine to yield urea, which is released into the blood and excreted by the kidney. Ornithine, the other product of the arginase reaction enters the mitochondrion in exchange for another molecule of citrulline via the same transporter.
Generally, substrate availability regulates the rate of the urea cycle; the higher the rate of ammonia production, the higher the rate of urea formation. N-acetyl-glutamate is an allosteric activator of carbamoyl phosphate synthetase I, and its synthesis is stimulated by arginine. During conditions of increased protein metabolism following ingestion of a high protein diet, or during fasting, when muscle protein is degraded to supply carbon skeletons for glucose production (gluconeogenesis), the urea cycle operates at an increased rate to eliminate excess nitrogen as urea. As fasting progresses, ketone body synthesis increases, diminishing the need for muscle protein breakdown to supply amino acids as a source of carbon skeletons for gluconeogenesis. This, in turn, decreases the need for increased nitrogen excretion as urea, and the urea cycle slows.
Deficiencies of the Urea Cycle
Deficiencies of the urea cycle are a threat to health because of the accumulation of ammonia, which is a neurotoxin. Normally free ammonia is fixed into either α-keto glutarate by glutamate dehydrogenase or glutamine by glutamine synthetase. The glutamine can be used by a variety of tissues to donate its amide nitrogen for the synthesis of nitrogen-containing compounds. The resulting glutamate donates its amino group, by transamination, primarily to pyruvate to form alanine, which carries the nitrogen to the liver. In the liver the nitrogen is removed from its carriers and fixed to carbamoyl phosphate by carbamoyl phosphate synthetase I, the first enzyme of the urea cycle.
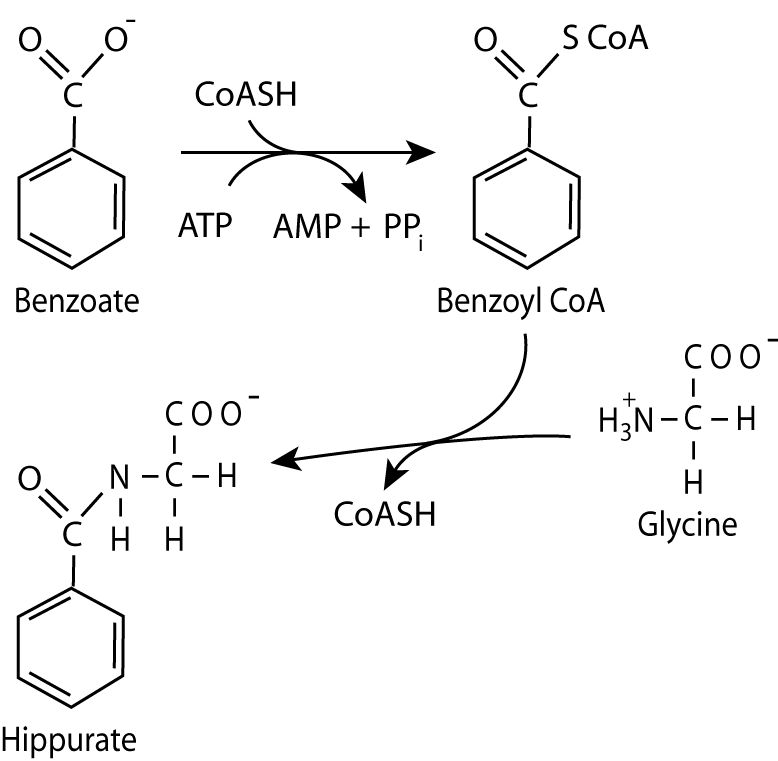
Benzoate, after conversion in the body to benzoyl CoA reacts with glycine, a non-essential amino acid, to form hippurate, which is excreted in the urine.
A urea cycle deficiency causes glutamine levels to increase, and because α-ketoglutarate is not regenerated by the removal of nitrogen from glutamine, the α-ketoglutarate level becomes too low to fix more free ammonia, which accumulates in the circulation.
The major clinical problem in treating patients with urea cycle deficiencies is to reduce the effects of excess ammonia on the nervous system, because high levels of ammonia are toxic to neurons, and cause irreversible neuronal damage.
The extent to which the elevation occurs depends on which enzyme of the urea cycle is deficient, and the key to treating a urea cycle deficiency is to identify the deficient enzyme.
The most common urea cycle deficiency is in ornithine transcarbamoylase (OTC), which is an X-linked disorder. It occurs with a frequency of 1/20,00 - 1/80,000 live births. The variation occurs because there is a late-onset form of OTC deficiency that may be underrepresented in the data used to determine the frequency of the deficiency in the population. Whatever the cause, a diet low in protein is essential to reduce the potential for excessive amino acid degradation with its associated generation of ammonia (ammonium ion).
Click The Image
Glycine Synthesis
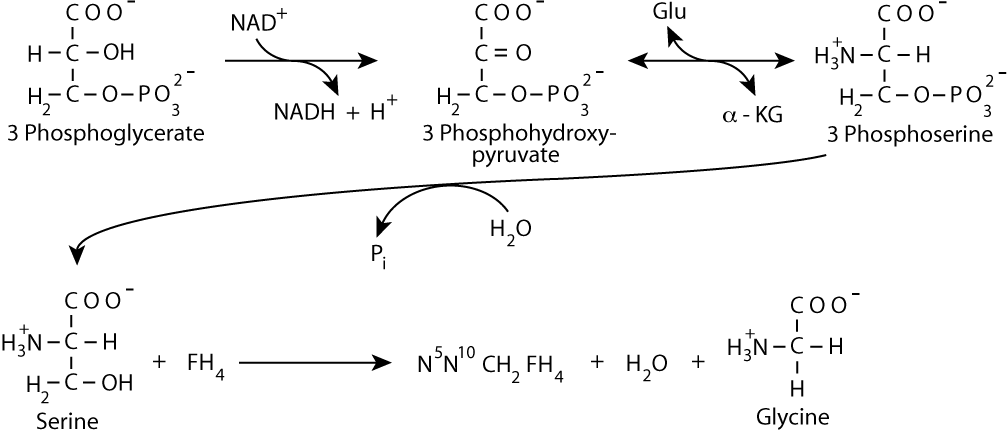
As glycine is converted to hippurate, which is excreted, the level of glycine in the body decreases. As a result, more glycine, a non-essential amino acid, is synthesized from 3 phosphoglycerate, requiring input of nitrogen as ammonia. [FH4 = tetrahydrofolate]
Glycine Synthesis
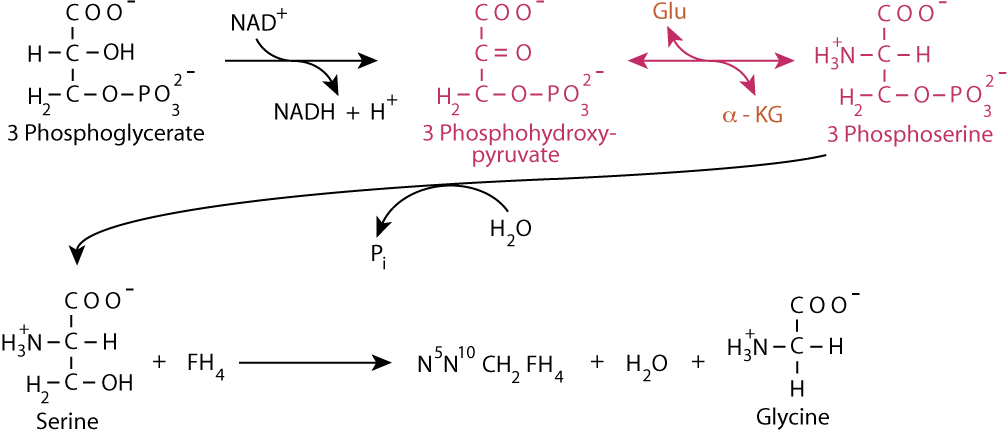
Glutamate donates the ammonia to 3 Phosphohydroxypyruvate via a transamination reaction, yielding 3 Phosphoserine and α-ketoglutarate. The α-ketoglutarate can then react, via either glutamate dehydrogenase or another transamination reaction to acquire another ammonia group, which it, in turn, can donate to another molecule of 3 Phosphohydroxypyruvate for the synthesis of another molecule of glycine, which can be eliminated from the body as hippurate. The repetitive resynthesis of glycine and its reaction with benzoyl CoA becomes the vehicle for the elimination of ammonia from the body in urine.
If the deficiency occurs before the synthesis of argininosuccinate, drugs that form conjugates with amino acids can be used for treatment. Benzoate (given as benzoic acid), after activation to benzoyl CoA, reacts with glycine to form hippurate, which is excreted. As a result, glycine is depleted, causing the body to synthesize more from 3 phosphoglycerate. In doing so it uses glutamate as a nitrogen donor in a transamination reaction, yielding α-ketoglutarate, which can then accept another nitrogen and continue in the synthesis of another molecule of glycine, which is conjugated to another molecule of benzoyl CoA for excretion as hippurate in repetitions of the cycle.
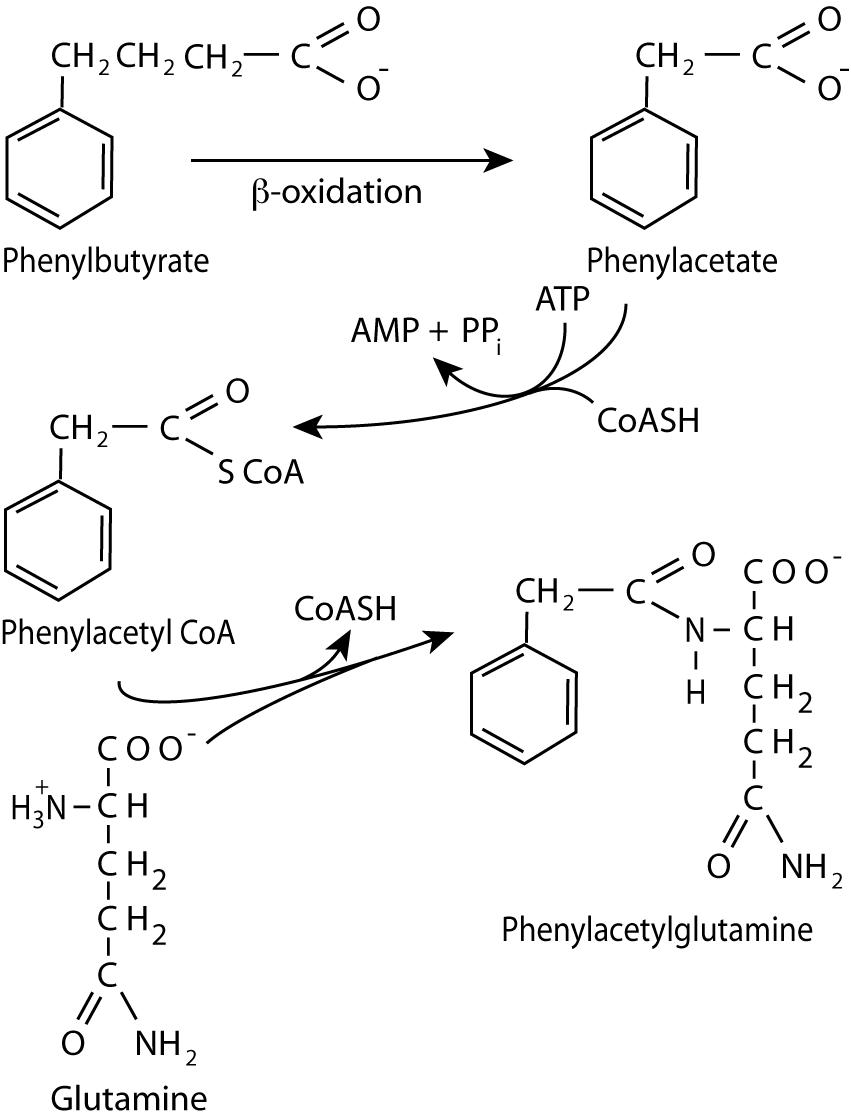
Phenylbuterate, after conversion in the body to phenylacetyl CoA reacts with glutamine, a non-essential amino acid, to form phenylacetylglutamine, which is excreted in the urine.
In addition, or alternatively, phenylbutyrate, can be used for treatment. It is converted to phenylacetate, the active compound, which conjugates with glutamine to form phenylacetylglutamine, which is excreted in the urine. Each molecule of phenylacetylglutamine excreted removes two nitrogens. As glutamine is depleted, the body synthesis more from glucose, first by synthesizing α-ketoglutarate and then converting it to glutamate either by transamination or the glutamate dehydrogenase reaction, and subsequently adding another nitrogen to the glutamate with glutamine synthetase, thereby using two nitrogens. As glutamine is a non-essential amino acid, as it is depleted, the body synthesizes more, and the cycle continues.
If the deficiency occurs after the synthesis of argininosuccinate large amounts of arginine may be beneficial. Once argininosuccinate has been synthesized, the two nitrogens destined for excretion have been incorporated in the substrate and the problem is that ornithine is not regenerated, causing it to be limiting. Ingesting large quantities of arginine leads to ornithine production by the arginase reaction and nitrogen excretion via argininosuccinate is enhanced.
Click The Image
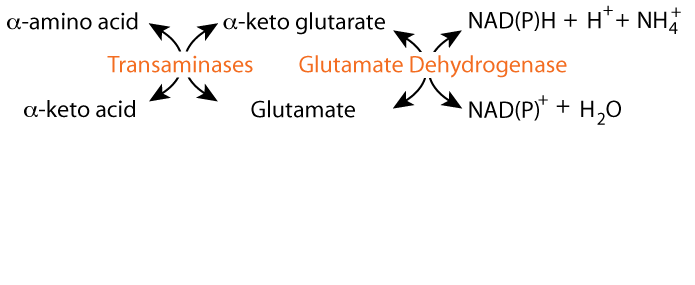
α-ketoglutarate can be converted to glutamate either by transamination, or by glutamate dehydrogenase. The glutamate dehydrogenase reaction fixes free ammonia (ammonium ion) and transamination reactions transfer ammonia from an amino acid. The resulting glutamate can donate its nitrogen to another α-keto acid by transamination, as in the formation of glycine, or ...
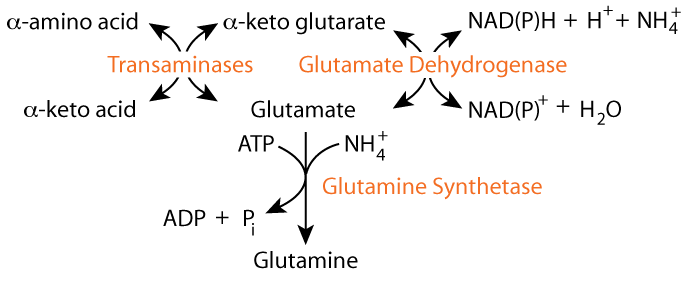
the glutamate can be converted to glutamine by glutamine synthetase, thereby using another free ammonium ion.
The severe hyperammonemia resulting from other urea cycle deficiencies rarely occurs in patients with arginase deficiency for at least two identifiable reasons: arginine can be released from the hepatocyte and excreted in urine since a second, inducible type II isozyme occurs in peripheral tissues, which can hydrolyze the arginine released by the hepatocyte to produce urea and ornithine The ornithine returns to the liver for use in the urea cycle, while the urea is excreted.
Because only a single tissue is involved, the liver, deficiencies in the urea cycle are good candidates for treatment by gene therapy since only one cell type, the hepatocyte, must be targeted by the vector that carries the replacement gene. Gene therapy experiments were carried out on individuals with ornithine transcarbamoylase deficiency, but were halted because one of the patients died of a severe immunologic reaction to the virus vector used to deliver the gene.