GLYCOGEN SYNTHESIS & DEGRADATION
 VI. Glycogen Synthesis.
VI. Glycogen Synthesis.
The liver is a so-called "altruistic" organ, which releases glucose into the blood to meet tissue need. Glucose released from muscle glycogen stores is used on site to provide energy for muscle contraction. Like glycolysis and gluconeogenesis, glycogenolysis and glycogenesis are NOT reversals of each other.
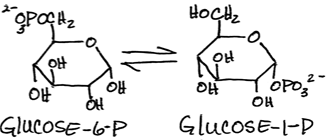 Interconversion of glucose-6-phosphate and glucose-1-phosphate by phosphoglucomutase:
Interconversion of glucose-6-phosphate and glucose-1-phosphate by phosphoglucomutase:
- reversible - this is the only enzyme common to both glycogen synthesis and breakdown
- Activation of glucose-1-phosphate to form UDP-glucose (uridine diphosphate-glucose) by UDPG pyrophosphorylase:
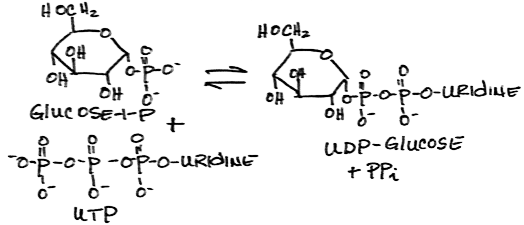
- Transfer of glucose from UDP-glucose to glycogen by glycogen synthase:
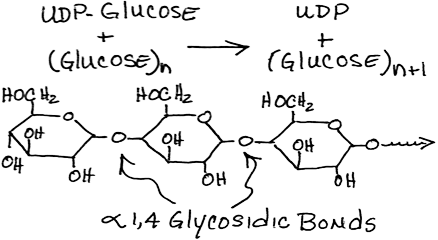
- does not initiate new molecules of glycogen but requires a "primer" at least 4 glucose units long
- Glycogenin, a protein, autocatalyzes the synthesis of a glycogen primer, covalently attached to itself, which glycogen synthase extends. Therefore, the number of molecules of glycogenin present determines the number of glycogen molecules synthesized.
- Glycogen synthase must always be in contact with glycogenin to be active. Therefore, the size of a glycogen molecule is limited by the physical distance between its most distal, non-reducing end and the glycogenin covalently attached to its reducing end. When this distance is longer than glycogen synthase can span, elongation of the glycogen molecule halts.
- inactivated by phosphorylation via cAMP-dependent signaling
- inactivation reversed by insulin, which stimulates dephosphorylation
- allosteric activation of the phosphorylated form by glucose-6-phosphate
- Transfer of (glucose)>6 from alpha-1,4 glycosidic linkage to 1,6 linkage by branching enzyme:
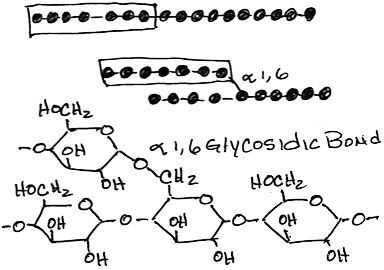
- more compact storage, more accessible free ends for synthesis and phosphorylase (see below)
- The block transferred is at least 7 residues long, must include the non-reducing end, must come from a chain of at least 11 residues long (thereby leaving a primer of at least 4 residues as is required by glycogen synthase) and is transferred at least 4 residues away from a pre-existing branch.
- Phosphorolysis of alpha-1,4-glycosidic bonds of glycogen to release glucose-1-phosphate sequentially from the non-reducing end by glycogen phosphorylase:
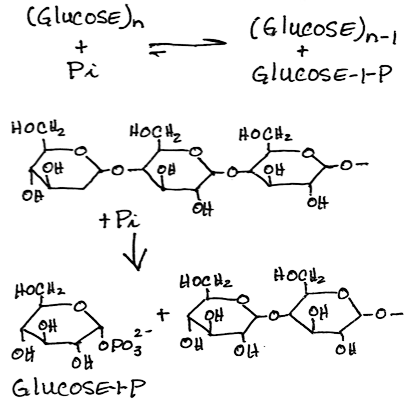 In vivo, [Pi] is about 100-fold higher than [glucose 1-phosphate], preventing reversal
In vivo, [Pi] is about 100-fold higher than [glucose 1-phosphate], preventing reversal- The muscle and liver phosphorylase isoforms are distinct.
- Glucose-binding causes a conformation change in the active phosphorylated liver "a" isoform, exposing an activating phosphate group to the enzymatic activity of protein phosphatase 1, which removes the activating phosphate, thereby regenerating the inactive, unphosphorylated "b" form of the enzyme. Thus, in liver, glycogen breakdown by glycogen phosphorylase is regulated by the glucose concentration, consistent with the function of the liver, to ensure that glucose is provided to other tissues.
- The muscle isoform of phosphorylase b can be activated by the binding of AMP, which is present in increased concentrations when the energy charge of the cell is low. ATP and glucose-6-phosphate reverse the activation by AMP by competing for the AMP binding site. Thus, in muscle, glycogen breakdown catalyzed by glycogen phosphorylase is regulated by the energy charge of the cell, consistent with the function of muscle glycogen to act as an energy store to be mobilized to supply energy for muscle contraction. The liver isoform of phosphorylase b is not activated by AMP.
- glycogen phosphorylase degrades linear glycogen in which the glucose units are linked by α 1,4 glycosidic bonds, to within 4 glucose units of an α 1,6 glycosidic branch, at which point glycogen phosphorylase stops
- Hydrolysis, not phosphorolysis, of glycogen branchpoints by transferase and alpha-1,6-glucosidase (debranching enzyme) to release glucose, not glucose 1-phosphate, from the α 1,6 glycosidic branch point:
- two step reaction, both activities in one protein
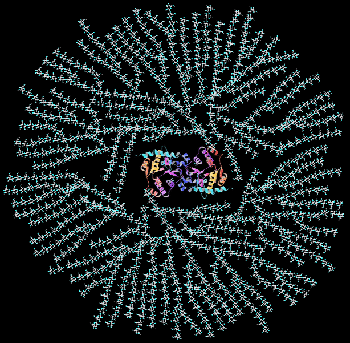 Cartoon of a glycogen granule: Glycogen is a highly branched molecule at the center of which is a dimer of the protein glycogenin to which the carbohydrate is covalently attached at a tyrosine residue in each monomer.
Cartoon of a glycogen granule: Glycogen is a highly branched molecule at the center of which is a dimer of the protein glycogenin to which the carbohydrate is covalently attached at a tyrosine residue in each monomer. VII. Glycogen Degradation.
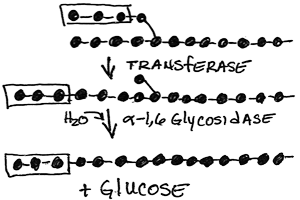
Reciprocal hormonal regulation of glycogen synthesis and glycogen degradation: Ensuring that glycogen synthesis and degradation do not occur simultaneously in a futile cycle
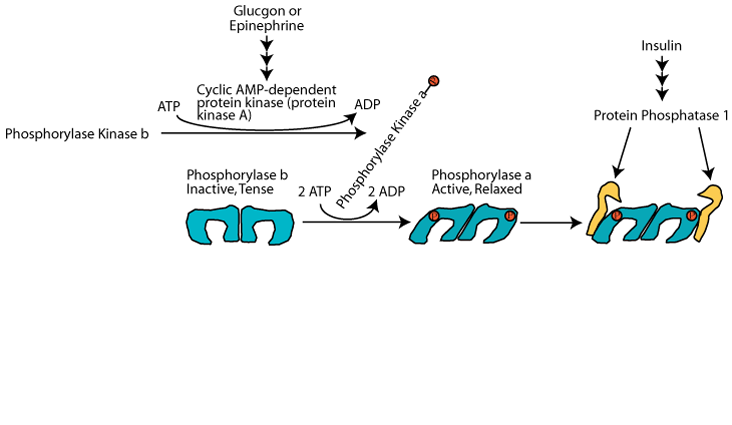
Glucagon (via glucagon receptors in liver) or epinephrine (via β2-adrenergic receptors in skeletal muscle and liver) triggered cAMP-dependent activation of Protein Kinase A (cAMP-dependent protein kinase) resulting in the phosphorylation of glycogen phosphorylase at serine 14, switching it from the unphosphorylated, inactive "b" form to the phosphorylated, activated, "a" form. However, protein kinase A does not phosphorylate glycogen phosphorylase directly. It phosphorylates Phosphorylase Kinase, converting it from the unphosphorylated, inactive "b" form" to the phosphorylated, active "a" form. Then the phosphorylated, active phosphorylase kinase phosphorylates glycogen phosphorylase at serine 14, converting it from the unphosphorylated, inactive b form to the phosphorylated, active a form. At the same time protein kinase A phosphorylates glycogen synthase. This has the effect of rendering the synthase susceptible to phosphorylation by several other protein kinases, which convert it from the unphosphorylated, active a form to the mutli-phosphorylated, inactive b form.
Click The Image
Reciprocal Hormonal Regulation of Glycogen Synthesis
and Degradation
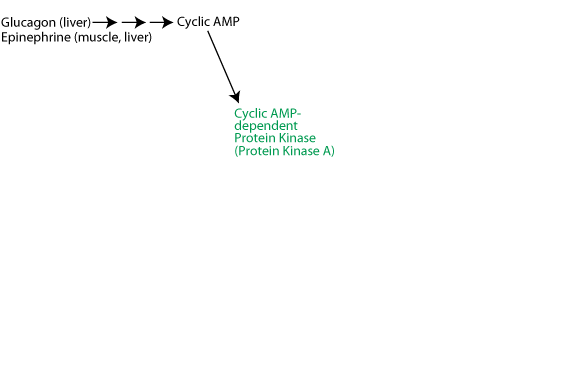
Activation of cyclic-AMP-dependent protein kinase: Glucagon (glucagon receptor in liver) or epinephrine (β2-adrenergic receptor in liver and muscle) signaling causes a rise in the intracellular concentration of cAMP, which activates
Reciprocal Hormonal Regulation of Glycogen Synthesis
and Degradation
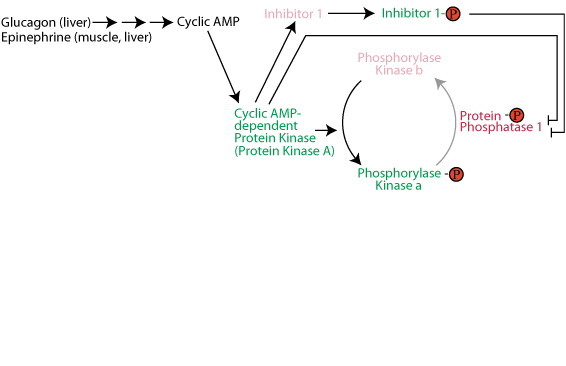
Activation of glycogen phosphorylase kinase and inhibition of protein phosphatase 1: Active cAMP-dependent protein kinase phosphorylates glycogen phosphorylase kinase (often referred to as simply phosphorylase kinase), converting it from the inactive "b" form to the active "a" form. Active cAMP-dependent protein kinase also phosphorylates the inhibitor 1 protein, activating it to bind to and inhibit protein phosphatase 1, and also phosphorylates protein phosphatase 1 directly, causing its inhibition. As a result the activating phosphate on phosphorylase kinase is not removed by protein phosphatase 1 and phosphorylase kinase remains active as long a glucagon or epinephrine signaling continues.
Reciprocal Hormonal Regulation of Glycogen Synthesis
and Degradation
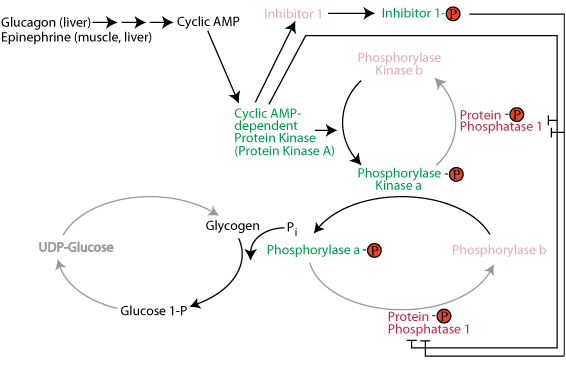
Activation of glycogen phosphorylase and phosphorolysis of glycogen: The active form of glycogen phosphorylase kinase phosphorylates and activates glycogen phosphorylase. Active Inhibitor 1 protein and direct phosphorylation by cAMP-dependent protein kinase keep protein phosphatase 1 in the inactive state so that is does not remove the activating phosphate group from glycogen phosphorylase. Active glycogen phosphorylase catalyzes the sequential removal of 1 glucose molecule as glucose 1-phosphate from the non-reducing ends of glycogen.
Reciprocal Hormonal Regulation of Glycogen Synthesis
and Degradation
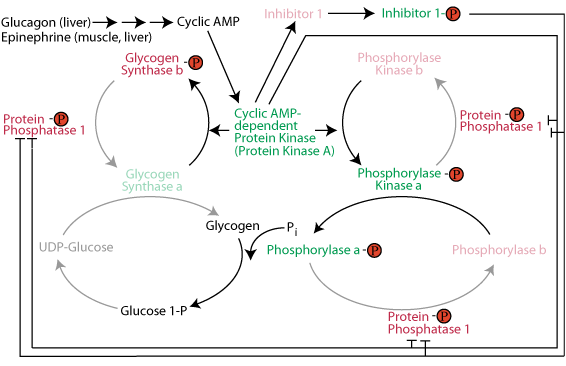
Inactivation of glycogen synthase: cAMP-dependent protein kinase phosphorylates glycogen synthase to make it susceptible to phosphorylation by other protein kinases that phosphorylate and inactivate it. The consequence is that when glycogen breakdown occurs, glycogen synthesis does not occur simultaneously in a futile glycogen breakdown/glycogen re-synthesis cycle. Active Inhibitor 1 protein and direct phosphorylation by cAMP-dependent protein kinase keep protein phosphatase 1 in the inactive state so that is does not remove the inactivating phosphate groups from glycogen synthase.
Reciprocal Hormonal Regulation of Glycogen Synthesis
and Degradation
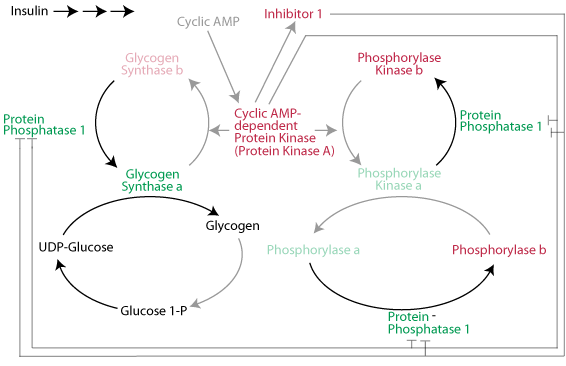
A rise in the level of blood glucose causes glucagon secretion by the pancreas to decline and the secretion of insulin to increase. The resulting decline in glucagon signaling (or a decrease in epinephrine signaling as a result of the resolution of an acute stress) causes cAMP-dependent protein kinase to become inactive. Insulin signaling causes the inhibitor 1 protein to become inactive and protein phosphatase 1 to become active. As a result, protein phosphatase 1 hydrolyzes the activating phosphate groups from glycogen phorphorylase kinase and glycogen phosphorylase and they both become inactive. At the same time protein phosphatase 1 hydrolyzes the inactivating phosphate groups from glycogen synthase and it becomes active. Glycogen synthesis proceeds and glycogen breakdown is prevented.
Glycogen storage diseases:
Imbalance between glycogenolysis and glycogenesis, or between branching and debranching activities results in storage of abnormal amounts of glycogen or of structurally abnormal glycogen, which can cause serious impairment of cell and organ functions. Human genetic defects in enzymes #3, #32 and #36-38 cause glycogen storage diseases. You should now be able to figure out for each mutant enzyme whether glycogen amount, structure, or both is affected. You should also consider, for enzymes with tissue-specific isomorphs, which organs may be affected. In addition to the enzymes described above, all cells contain a lysosomal enzyme, alpha 1,4-glycosidase. Mutations in its gene have global effects.
Question:
What clinical characteristics do you expect from a defect in phosphorylase kinase?Phosphorylase kinase (PhK) deficiency causing glycogen storage disease type IX (GSD IX) results from deficiency of the enzyme phosphorylase b kinase, which has a major regulatory role in the breakdown of glycogen. The two types of PhK deficiency are liver PhK deficiency characterized by early childhood onset of hepatomegaly and growth restriction, and often, but not always, fasting ketosis and hypoglycemia, and muscle PhK deficiency, which is considerably rarer and characterized by any of the following: exercise intolerance, myalgia, muscle cramps, myoglobinuria, and progressive muscle weakness. Although symptoms and biochemical abnormalities of liver PhK deficiency were thought to improve with age, it is becoming evident that patients need to be monitored for long-term complications such as liver fibrosis and cirrhosis.
Epinephrine activates α1 adrenergic receptors in the liver to regulate glycogen synthesis and breakdown. Epinephrine signaling via the α1 adrenergic receptor activates glycogenolysis and inhibits glycogen synthesis, mainly by increasing hepatocyte Ca2+ levels. The effects of epinephrine at the α1 adrenergic receptor are mediated by phosphatidylinositol bisPhosphate (PIP2)-Ca2+ signal transduction, a principal intracellular second messenger signaling system used by many hormones.
The signal is transferred from the epinephrine receptor to membrane-bound phospholipase C by a Gq protein. Phospholipase C hydrolyzes PIP2 to form diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 stimulates the release of Ca2+ from the endoplasmic reticulum. Ca2+ and DAG activate protein kinase C, and Ca2+ binds to the clacium-binding protein calmodulin.
Calcium/calmodulin associates as a subunit with several enzymes and modifies their activities. It binds to inactive phosphorylase kinase, partially activating it — the fully active enzyme is both bound to the calcium/calmodulin and phosphorylated. Phosphorylase kinase phosphorylates glycogen phosphorylase b, thereby activating glycogen degradation. Calcium/calmodulin is an activator of one of the glycogen synthase kinases (calcium/calmodulin synthase kinase). Protein kinase C, calcium/calmodulin synthase kinase, and phosphorylase kinase all phosphorylate glycogen synthase, which inhibits it.
Epinephrine in the liver enhances or is synergistic with the effects of glucagon. Epinephrine release during hypoglycemia or exercise can stimulate hepatic glycogenolysis and inhibit glycogen synthesis very rapidly.
Click The Image
Hepatocyte α1 adrenergic receptor:
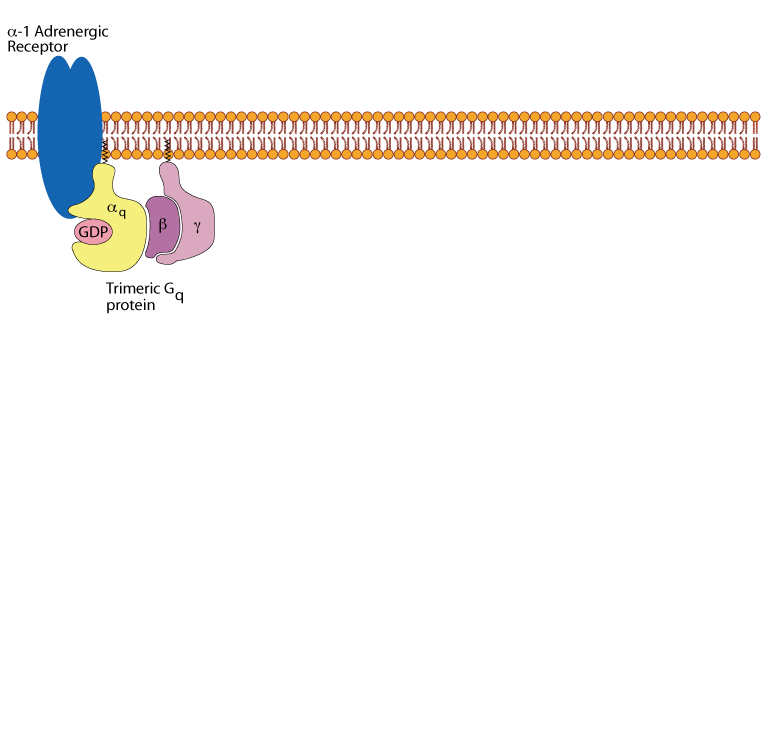
α1 adrenergic receptors transmit their signals to a trimeric Gq protein.
Hepatocyte α1 adrenergic receptor: epinephrine binding
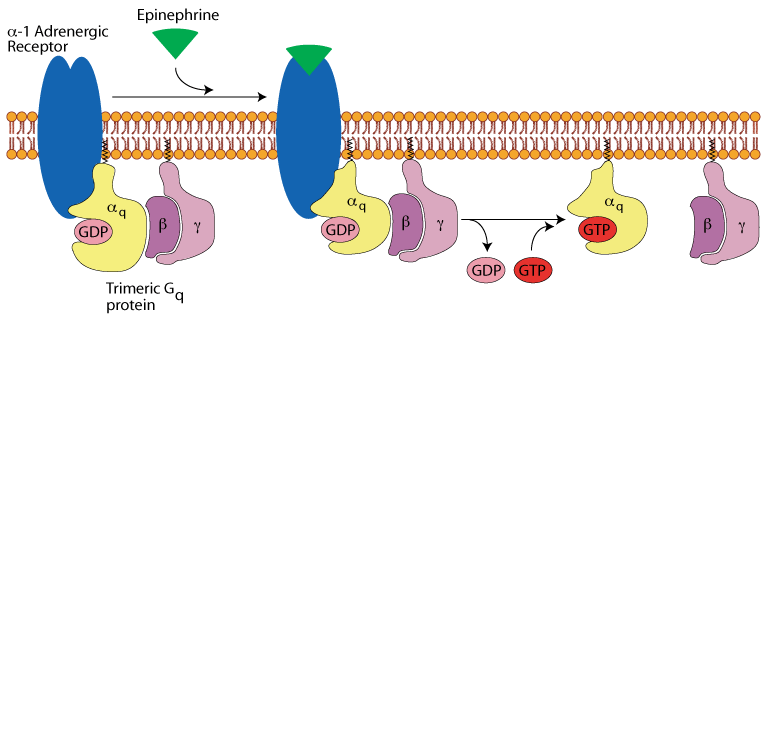
Binding of epinephrine to the α1 adrenergic receptor transmits its signal to a trimeric Gq protein, which is activated by exchanging its bound GDP for GTP. It dissociates into the active α subunit and the β/γ subunit pair.
Hepatocyte α1 adrenergic receptor: activation of phospholipase C
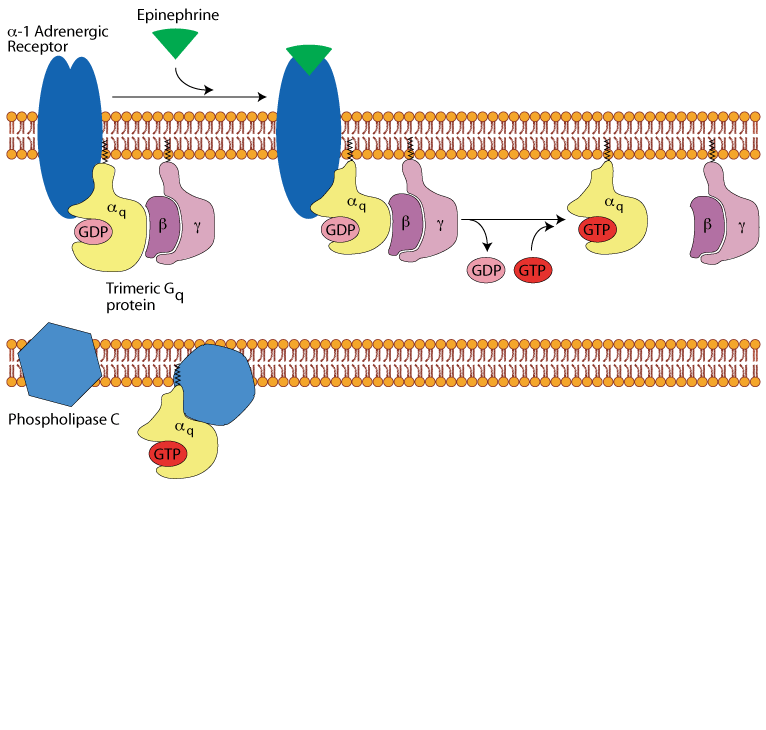
The active, GTP-bound, Gq alpha subunit binds to and activates phospholipase C, which
Hepatocyte α1 adrenergic receptor: diacyl glycerol & inositol tris-phosphate
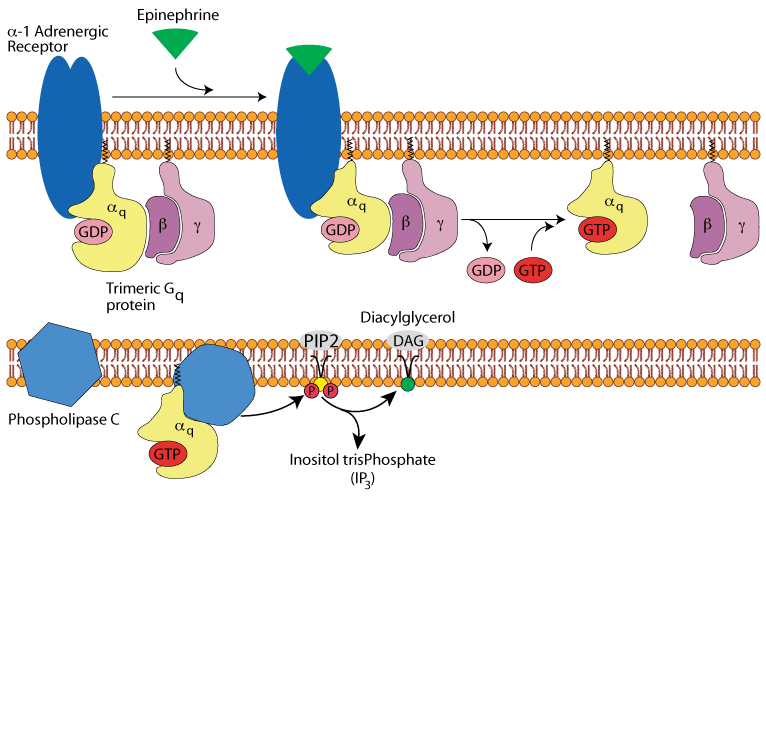
hydrolyzes phosphoinositol bisPhosphate (PIP2), converting it to diacylglycerol (DAG) and inositol trisPhosphate (IP3).
Hepatocyte α1 adrenergic receptor: phospholipase C reaction
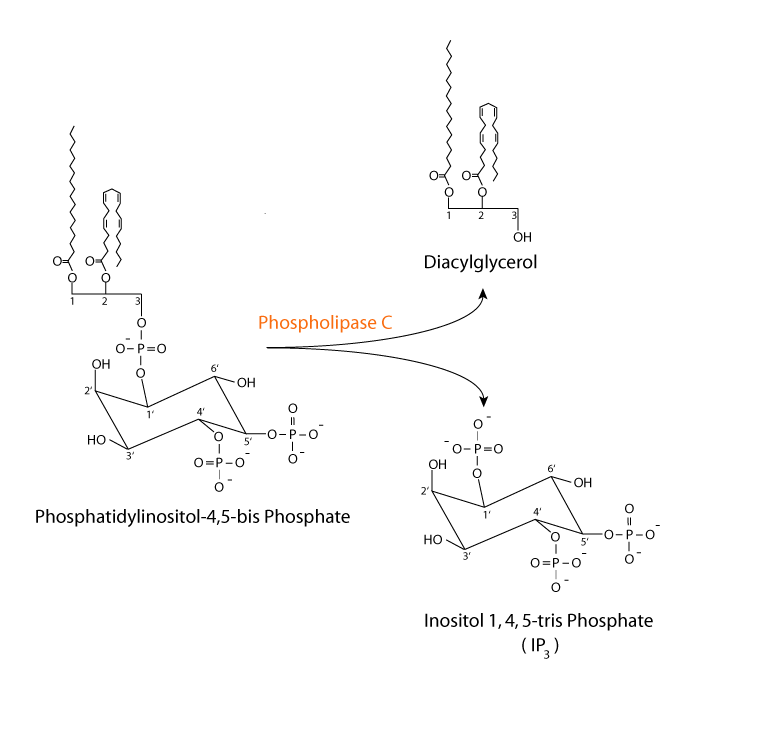
Hepatocyte α1 adrenergic receptor: calcium & diacyl glycerol
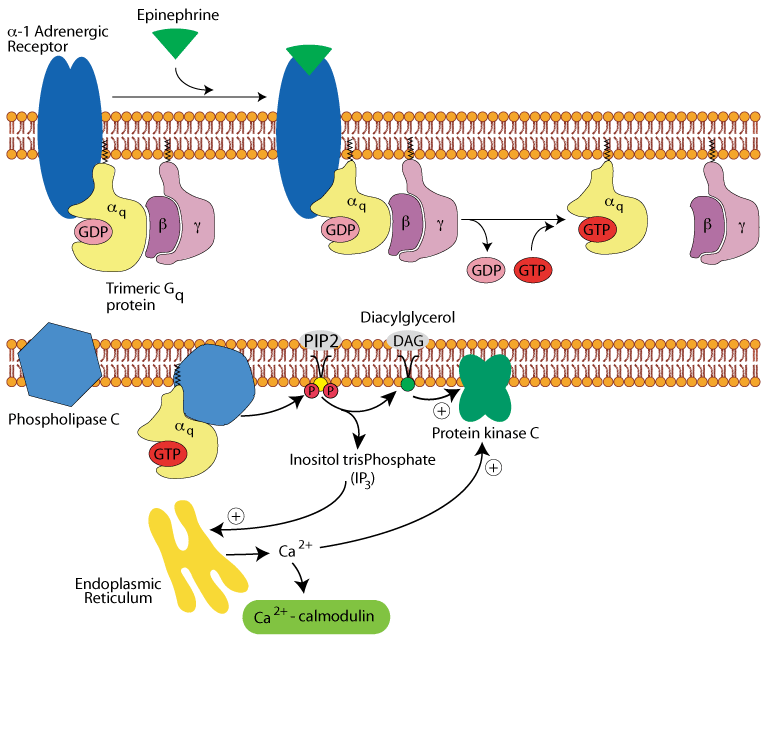
IP3 acts on the endoplasmic reticulum to release stored Ca2+. The released Ca2+ and DAG activate protein kinase C. The Ca2+ binds to and activates calmodulin.
Hepatocyte α1 adrenergic receptor: activation of two more protein kinases
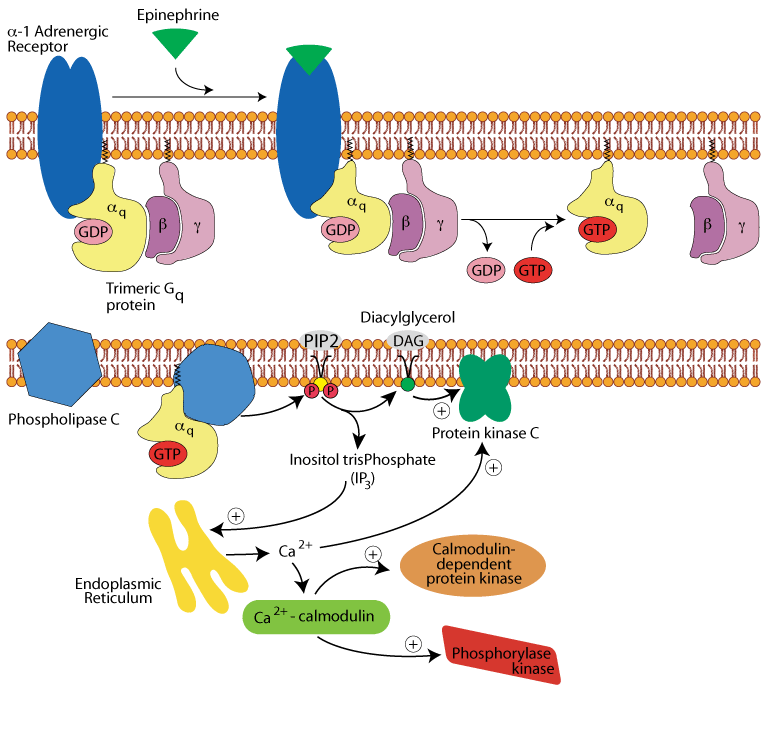
Calcium/calmodulin bind to and active calmodulin-dependent protein kinase and glycogen phosphorylase kinase.
Hepatocyte α1 adrenergic receptor: glycogen synthase & phosphorylase
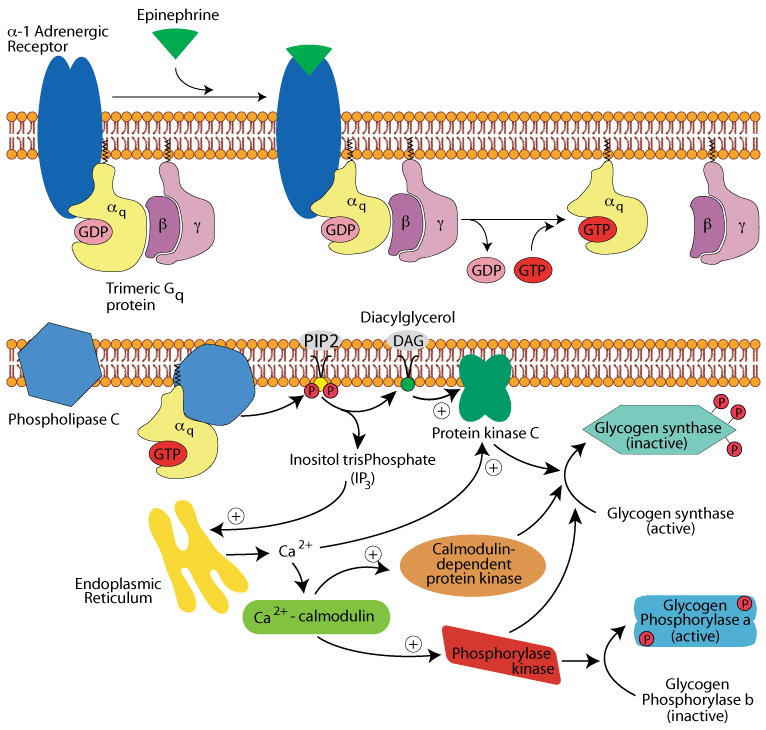
All three activated protein kinases phosphorylate and inactivate glycogen synthase. Glycogen phosphorylase kinase phosphorylates inactive glycogen phosphorylase b, converting it to active, glycogen phosphorylase a. Glycogen synthesis (glycogenesis) is inhibited and glycogen breakdown (glycogenolysis) is activated.
Liver glycogen increases after a meal, is used as the major source of glucose during a short fast or early in a longer fast, but is consumed after a fast of approximately 20 -30 hours.
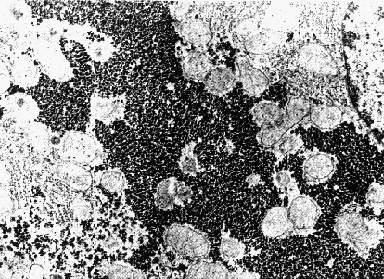 Electron micrograph of the liver of a well-fed rat. The dark granular material is composed of glycogen granules. The nucleus with darkly-staining chromatin at the nuclear membrane is visible at the upper right.
Electron micrograph of the liver of a well-fed rat. The dark granular material is composed of glycogen granules. The nucleus with darkly-staining chromatin at the nuclear membrane is visible at the upper right. 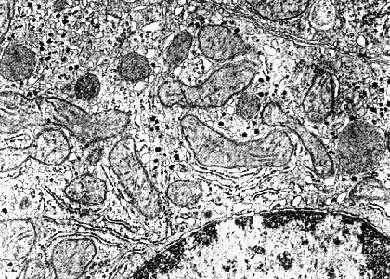 Electron micrograph of the liver of a 24-hour fasted rat. Only a few dark glycogen granules remain. The nucleus with darkly-staining chromatin at the nuclear membrane is visible at the lower right.
Electron micrograph of the liver of a 24-hour fasted rat. Only a few dark glycogen granules remain. The nucleus with darkly-staining chromatin at the nuclear membrane is visible at the lower right. The different liver and muscle isoforms of glycogen phosphorylase are regulated according to the tissue-specific function of glycogen stored in each tissue.
Degradation of liver glycogen is triggered by activated Protein Kinase A in response to increased cyclic-AMP resulting from either glucagon or epinephrine signaling. As Protein Kinase A phosphorylates the liver bifunctional enzyme, thereby causing a reduction in the activity of Phosphofructokinase 2 and an increase in the activity of Fructose 2,6-bisPhosphatase, the activity of Phosphofructokinase 1 is reduced. In addition, Protein Kinase A inactivates Pyruvate Kinase by phosphorylation. The result is that the glucose phosphate generated by the degradation of liver glycogen, instead of entering the liver glycolytic pathway, is dephosphorylated and the resulting glucose is released into the blood.
Click The Image
Inactive Liver Glycogen Phosphorylase b

When not phosphorylated, liver glycogen phosphorylase, which is a homodimer, is in the tense, inactive b form.
Activation by phosphorylase kinase
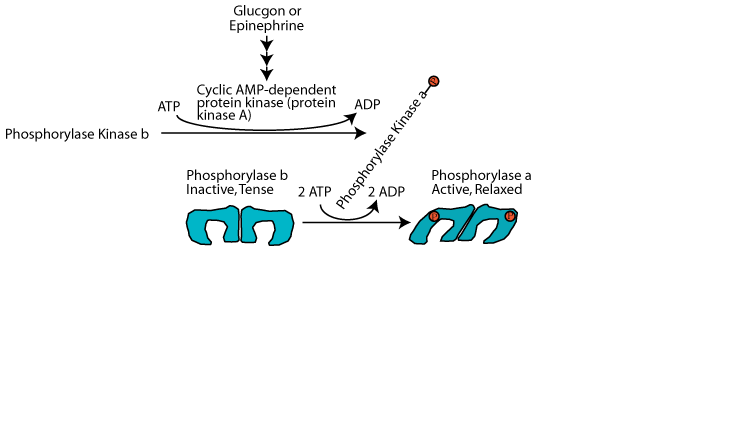
In response to glucagon signaling glycogen phosphorylase kinase is activated by phosphorylation by cAMP-dependent protein kinase (protein kinase A) and it in turn phosphorylates glycogen phosphorylase on each monomer, converting it from the tense inactive b form to the relaxed, active a form.
Binding of Protein Phosphatase 1
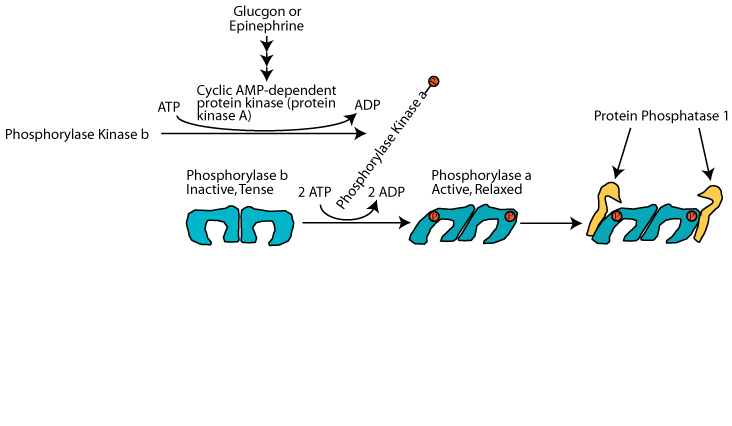
Protein phosphatase 1 can bind to active glycogen phosphorylase, but it is inactive during glucagon or epinephrine signaling.
Switch from Glucagon Signaling to Insulin Signaling
When the level of glucose raises sufficiently, glucagon secretion from the pancreas is decreased and insulin secretion is increased. The increased insulin causes activation of protein phosphatase 1 but the phosphate groups on glycogen phosphorylase are positioned so that they are inaccessible to the protein phosphatase 1.
Glucose binding causes the activating phosphates to become accessible
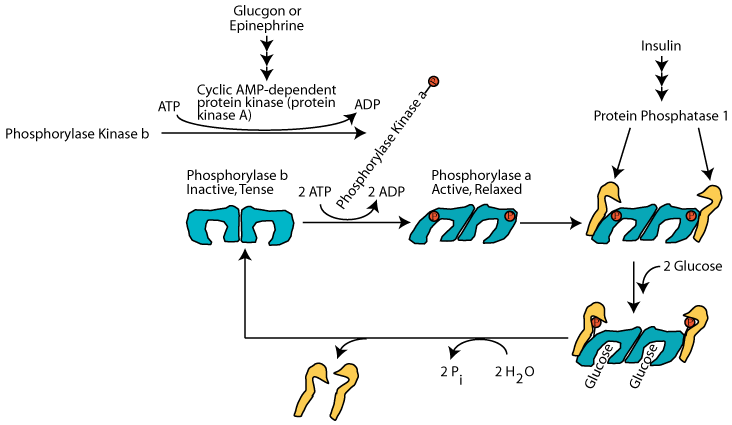
Only when the glucose level has risen sufficiently does glucose bind to each subunit of active glycogen phosphorylase, causing a conformation change that makes the activating phosphates accessible to active protein phosphatase 1, which hydrolyzes them and converts glycogen phosphorylase to the inactive, tense b form. The phosphatase does not bind to inactive glycogen phosphorylase b, but rather is freed to hydrolyze the inactivating phosphates from glycogen synthase, thereby activating it. Glucose does not regulate its activity of muscle glycogen phosphorylase.
Skeletal muscle glycogen phosphorylase is regulated by the availability of cellular energy. An increase in AMP concentration, which occurs during strenuous exercise, signals energy demand. As AMP increases it binds to and allosterically increases the activity of skeletal muscle glycogen phosphorylase, thereby increasing glycogen degradation to glucose 1-phosphate. Phosphoglucomutase converts the glucose 1-phosphate to glucose 6-phosphate, which supplies the glycolytic pathway (and subsequently, the TCA cycle and electron transport chain) with substrate for the generation of ATP energy. Increased glucose 6-phosphate and ATP reverse the allosteric activation of glycogen phosphorylase by AMP.
Ca2+ released from the skeletal muscle sarcoplasmic reticulum during muscle contraction activates calmodulin, which activates phosphorylase kinase, which phosphoylates glycogen phosphorylase, which degrades glycogen to produce glucose phosphate to supply energy for muscle contraction.
Skeletal muscle glycogen is also regulated hormonally by epinephrine signaling through β2 adrenergic receptors to triggor glycogen degradation in response to acute stresses, but it is not regulated by glucagon, as skeletal muscle does not have glucagon receptors. As the muscle bifunctional enzyme and pyruvate kinase lack phosphorylation sites, the glucose phosphate generated in response to epinephrine signaling is directed into the glycolytic pathway to supply energy for muscle to react to the extant acute stress.
Click The Image
Inactive Muscle Glycogen Phosphorylase b

When not activated, muscle glycogen phosphorylase, which is a homodimer, is in the tense, inactive b form.
Allosteric Activation By AMP
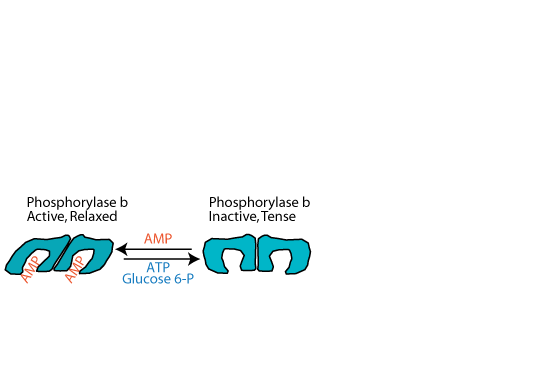
As ATP is used for energy, first ADP and then AMP accumulates. As the level of AMP rises, AMP activates muscle glycogen phosphorylase by binding to it and converting it to the relaxed state. This activation does not require phosphorylation of the glycogen phosphorylase, and therefore it is referred to as the "b", rather than the "a" form. When ATP or glucose-6-phosphate, ready sources of energy, increase, they compete for binding with AMP and convert glycogen phosphorylase back to the inactive, tense state. Liver glycogen phosphorylase does not bind, and is not activated by AMP.
Activation By Muscle Contraction
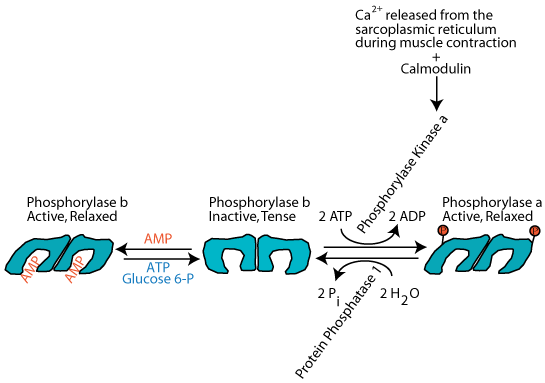
Nerve impulses that cause muscle contraction induce the release of Ca2+ from the sarcoplasmic reticulum. This Ca2+ activates calmodulin, a modifier protein that binds to glycogen phosphorylase kinase and converts it to the active form without it becoming phosphorylated. The activated phosphorylase kinase phosphorylates glycogen phosphorylase, converting it to the active, relaxed "a" form. The active glycogen phosphorylase catalyzes the phosphorylation of glycogen to supply glucose as a source of energy for the ensuing muscle contraction.
Activation By Phosphorylase Kinase
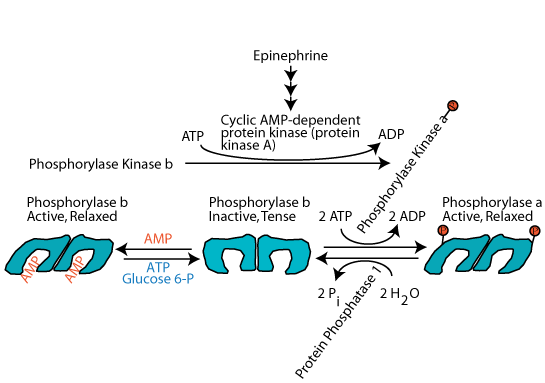
Epinephrine, the major acute stress hormone that signals though the β-2 adrenergic receptor, a G-protein coupled receptor, induces the activation of muscle cell adenylyl cyclase, which, in turn, activates cAMP-dependent protein kinase (protein kinase A), which phosphorylates and activates glycogen phosphorylase kinase. The active glycogen phosphorylase kinase then phosphorylates glycogen phosphorylase, converting it from the inactive, tense, b form" to the active, relaxed, a form. By this mechanism, epinephrine induces the breakdown of muscle glycogen to supply muscle cells with increased glucose as a ready source of energy to respond to an extant acute stress.
| Approximate Distribution of Body Fuels and Their Potential Energy in a 70-kg Adult Human | ||
| Kilograms | Kilocalories | |
| Adipose tissue triacylglycerol | 12 | 108,000 |
| Muscle protein (dry weight) | 6 | 24,000 |
| Carbohydrate (dry weight) Muscle glycogen Liver glycogen |
0.3 0.1 | 1,200 400 |
| Triacylglycerols are a more efficient form of energy storage | |
| Triacylglycerols: Protein: Carbohydrate: |
9 Kilocalories/gram 4 Kilocalories/gram 4 Kilocalories/gram |
| The three fatty acids in triacylglycerols are more reduced than either carbohydrates or protein carbon skeletons. Therefore, the oxidation of fatty acids yields more electrons per gram to drive the production of ATP energy than either carbohydrates or protein carbon skeletons.
Each gram of stored glycogen is hydrated with approximately 3 grams of water. 108 Kilograms of glycogen + its associated water would be required to store as many calories as are stored by 12 Kilograms of triacylglycerols in a 70 kg adult human. |
|
Carbohydrate fuel is stored as glycogen in the muscles and liver, approximately 75% in the muscles and 25% in the liver. Approximately 3 grams of water are required to hydrate each gram of stored glycogen.
Triacylglycerol StorageTriacylglycdrols (fats) are stored in adipose tissue located in almost all parts of the body. Significant fat is stored under the skin (subcutaneous) and in adipose tissue. Other storage sites are abdomen (central adiposity), buttocks, thighs and upper arms (peripheral adiposity). Fat storage location is determined by genetics and individual hormone balance. Testosterone favours central adiposity, and estrogen causes fat storage peripherally.
Central adiposity is considered more of a risk factor for coronary heart disease than peripheral storage, and it is thought that this is because fat contained in the peritoneal cavity (central obesity) is significantly more likely to be linked to metabolic disturbances such as insulin resistance and elevated plasma lipids.
Protein StorageProtein is not stored in the same way as carbohydrate and triacylglycerols. Other than around 100g in the amino acid pool, protein is not stored. It forms muscle and organ tissue, so it is mainly used as a building material rather than an energy store. However, tissue proteins are broken down to release energy during fasting/starvation, so muscles and organs do represent a large potential energy store, but protein degradation to provide carbon skeletons for energy production occurs at the expense of tissue structure and function.
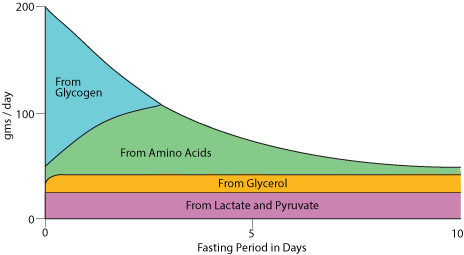
Glucose release from the liver during fasting / starvation. The fast begins (at time "0" in this diagram) when all food that has been eaten is cleared from the stomach and small intestine.
A fast begins approximately between four and eight hours after the last meal (depending on the size of the meal), when food has been cleared from the stomach and small intestine.
As the entry of food molecules into the blood from the small intestine slows, liver glycogen is degraded and is the main source of blood glucose. At the same time, adipose tissue triacylglycerol stores (fat) begin to be hydrolysed to fatty acids and glycerol. The fatty acids provide tissues that have mitochondria (the site of fatty acid β oxidation) with an alternate source of energy, and the glycerol is largely used by the liver for the synthesis of glucose from non-carbochdrate carbon skeletons. Lactate, ionized lactic acid produced largely from the reduction of pyruvate from extra-hepatic tissues, sypplies carbon skeletons for gluconeogenesis. Proteins in extrahepatic tissues are degraded to provide amino acids whose carbon skeletons are used by the liver for gluconeogenesis. The protein nitrogen is converted to urea by the liver for excretion by the kidney. The liver uses some of the fatty acids for the synthesis of ketone bodies, which can be osidized for energy by tissues that have mitochondria. After approximately 24-48 hours liver glycogen is exhausted and gluconeogenesis is the sole source of blood glucose. As the fast continues, the blood [glucose] declines and evertually reaches a constant concentration maintained exclusively by gluconeogenesis. This lower blood [glucose] serves those tissues that are unable to used fatty acids as a source of energy, mainly red blood cells (RBC) and the brain. RBC have no mitochondria and, therefore, can't use either fatty acids or ketone bodies as a source of energy. The brain has mitochondria and replaces approximately 80% of its glucose requirement with ketone bodies. The brain does not used fatty acids as a major source of energy because the transport of fatty acids across the blood-brain barrier is too slow.