BRANCHED-CHAIN AMINO ACIDS
Unlike most other amino acids, which are degraded mainly in the liver, the branched-chain amino acids — isoleucine, leucine and valine — are degraded predominantly in extra-hepatic tissues, mainly in muscle, because extra-hepatic tissues have higher activities of the transaminases for the branched-chain amino acids and of the enzyme branched-chain α-ketoacid dehydrogenase, the second enzyme in the degradation pathways for branched-chain amino acids, than does the liver. Normally, the nitrogen from branched- chain amino acids is transported to the liver for removal as urea, but in some physiological states it is diverted for use by other tissues. Oxidation of branched-chain amino acids in muscle serves two functions. The first is the generation of ATP, and the second is the synthesis of glutamine, which effluxes from the muscle. The highest rates of branched-chain amino acid oxidation occur under conditions of acidosis, in which there is a higher demand from glutamine to transfer ammonia to the kidney and to buffer the urine as ammonium ion during proton excretion (see “AA Flux” in the top menu). The degradation of isoleucine and valine provides a source of energy and a source of precursors to replenish TCA cycle intermediates (anaplerosis). Both amino acids degrade to produce succinyl CoA, a TCA cycle intermediate; isoleucine degradation also yields acetyl CoA . Leucine degradation yields acetoacetate, a ketone body, and acetyl CoA, both of which are a source of energy.Click The Image
Transamination
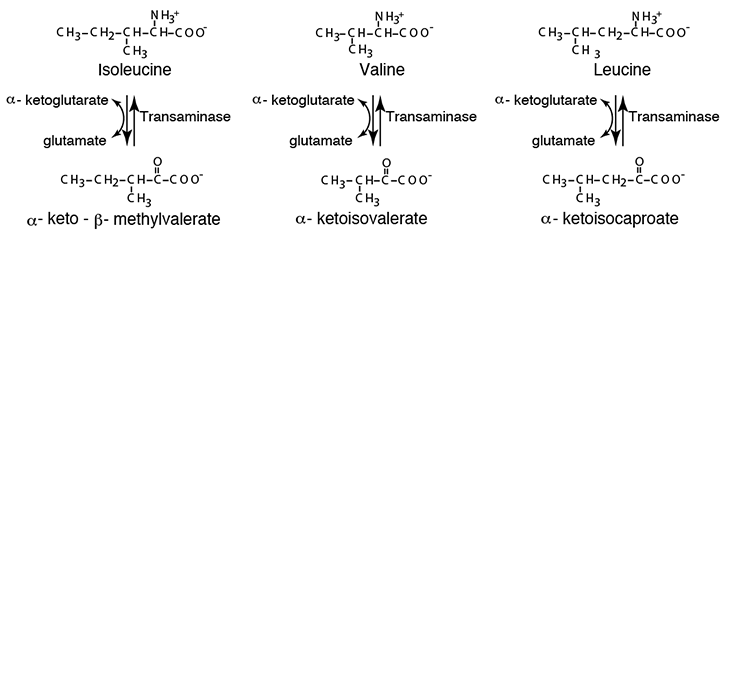
The first step in the degradation of the branched-chain amino acids is transamination to remove the amino nitrogen. A specific transaminase exists for each of the three branched-chain amino acids. As for all transaminases, α-ketoglutarate accepts the amino acid amino group, yielding glutamate and the specific cognate α-ketoacid of each branched-chain amino acid.
Branched-Chain α-Ketoacid Dehydrogenase
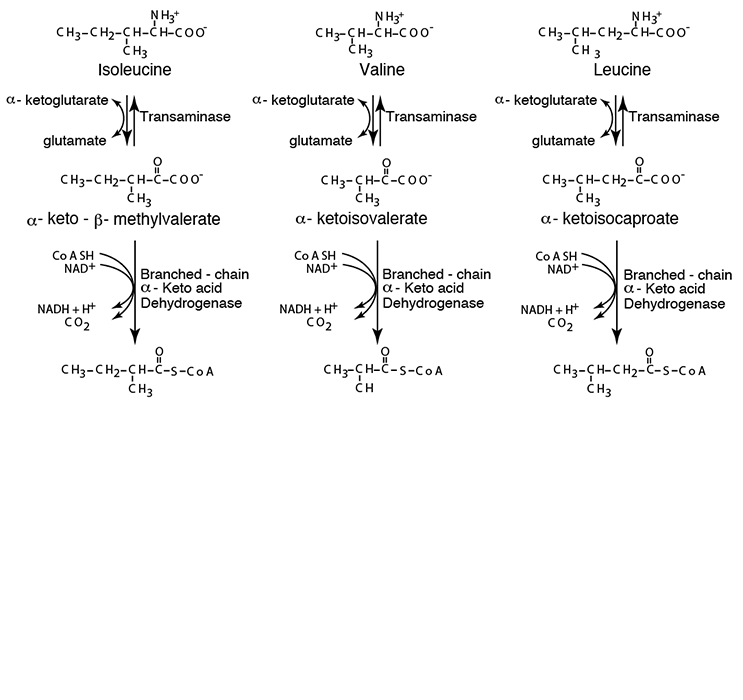
Each of the three cognate α-ketoacids undergoes oxidative decarboxylation by a single α-Ketoacid Dehydrogenase similar to Pyruvate Dehydrogenase and α-Ketoglutarate Dehydrogenase.
Propionyl CoA —> Succinyl CoA, Acetyl CoA, and Acetoacetate
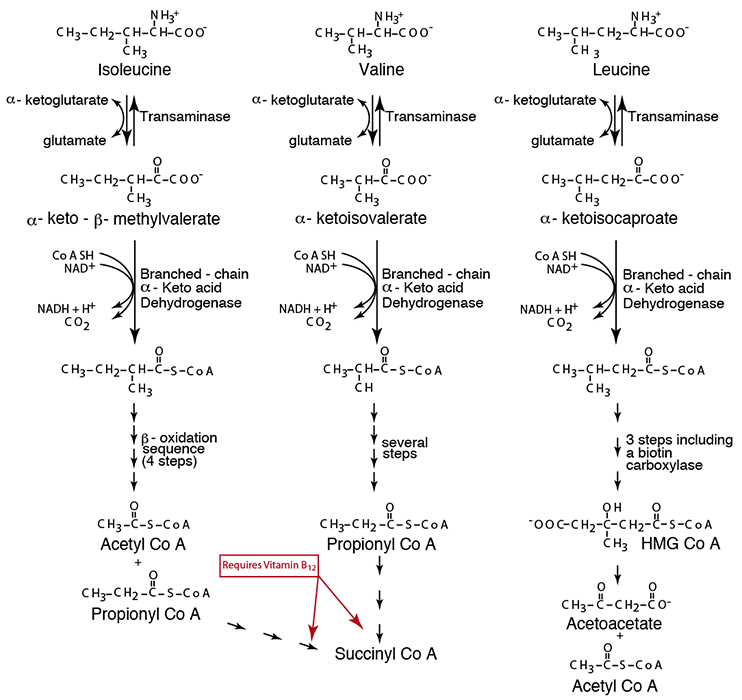
After a few more enzymatic reactions (not specifically indicated here), each branched-chain amino acid carbon skeleton yields it final product — Isoleucine yields Acetyl CoA and Propionyl CoA, Valine yields Propionyl CoA, and Leucine yields Acetyl CoA and Acetoacetate. Propionyl CoA is converted by a three-step reaction series that requires vitamin B12 (see also “Synthesis & Degradation > Methionine” in the top menu) and enters the TCA Cycle, which converts it into other TCA Cycle intermediate, one of which is α-Ketoglutarate that can be converted to Glutamine for efflux from muscle (see “AA Flux” in the top menu).
- transamination of the branched-chain amino acids isoleucine, valine and leucine, by Transaminases specific for each, to form the cognate α-ketoacids, α-keto-β-methylvalerate, α-ketoisovalerate and
α-ketoisocaproate, respectively
- the branched-chain amino acids play a special role in muscle and most other tissues because they are the major amino acids which can be oxidized in tissues other than liver; brain, heart, kidney and skeletal muscles have high activity of branched-chain amino acid transaminase relative to liver
- oxidative decarboxylation of each resulting α-ketoacid by a single enzyme complex, Branched-chain α-ketoacid dehydrogenase, in mitochondria
- branched-chain α-ketoacid dehydrogenase generates the CoA thiol ester derivative from each of the branched-chain α-ketoacids, which then follow specific oxidative (NADH- and FADH2 yielding) pathways to yield their ultimate CoA thiol ester derivatives. Isoleucine yields acetyl CoA, which is a ketone body precursor, and propionyl CoA, which is glucogenic via succinyl CoA. Valine yields propionyl CoA, which is glucogenic. Leucine yields acetoacetate, which is a ketone body, and acetyl CoA, which is ketogenic. The conversion pathway from propionyl CoA to succinyl CoA requires vitamin B12 and biotin (vitamin B7).
Generalized Structure of α-Keto Acid Dehydrogenase Complexes
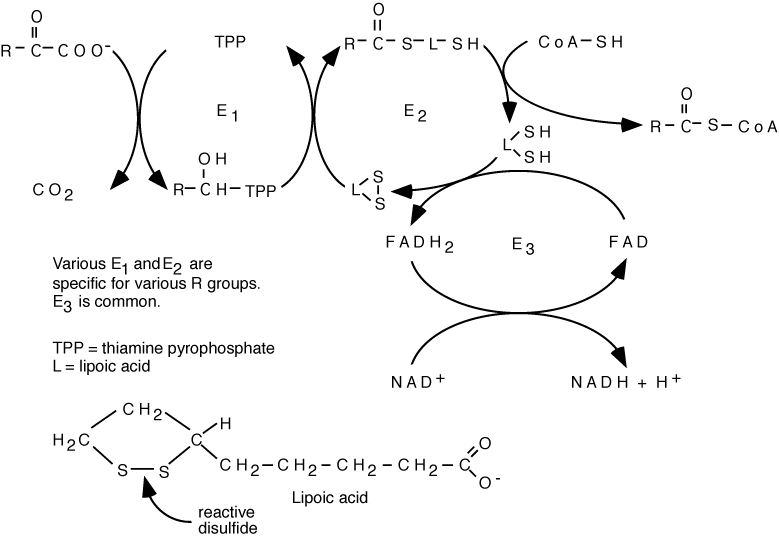
The three α-Ketoacid Dehydrogenases, Pyruvate Dehydrogenase, α-Ketoglutarate Dehydrogenase, and Branched-Chain α-Ketoacid Dehydrogenase all have the same general structure. The E1 and E2 components are specific for their specific substrates, the E3 component is common to all three dehydrogenases.
Three different α-ketoacid dehydrogenases have a similar subunit structure. However, the E1 and E2 components are substrate-specific (each of the components has a specific name that reflects its specific enzymatic function, but generally the three components are referred to as E1, E2, or E3), i.e., they recognize different R groups. Pyruvate Dehydrogenase recognizes pyruvate, converting it to acetyl CoA; α-Ketoglutarate Dehydrogenase recognizes α-ketoglutarate, converting it to succinyl CoA; Branched-chain α-Ketoacid Dehydrogenase recognizes all three branched-chain α-ketoacids generated from the three branched-chain amino acids by their respective, specific transaminases. A deficiency in the common E3 component of the enzyme complexes affects pyruvate dehydrogenase, α-ketoglutarate dehydrogenase and branched-chain α-ketoacid dehydrogenase because it is shared among them, while deficiencies in either the E1 or the E2 component of the complexes affects only the pathway for which it is specific, i.e., either pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, or branched-chain α-ketoacid dehydrogenase.
The α-Ketoacid Dehydrogenases required several prosthetic groups: E1 requires vitamin B1 (thiamin), E2 requires lipoic acid (not a vitamin, is synthesized in humans) and vitamin vitamin B5 (pantothenate, for Coenzyme A), E3 requires vitamin B2 (riboflavin for FAD) and vitamin B3 (niacin for NAD)
- Skeletal muscle branched-chain α-ketoacid dehydrogenase activity is increased by cortisol signaling, or when amino acids concentrations are high as, for example, following a high protein meal.
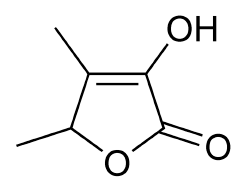
Sotolon (4,5-dimethyl-3-hydroxy-2[5H]-furanon4,5-dimethyl-3-hydroxy-2[5H]-furanone) is thought to be the source of the maple syrup odor in Branched Chain Keto Acid Dehydrogenase defiency (Maple Syrup Urine Disease). Sotolon (also known as sotolone) is a lactone and an extremely powerful aroma compound, with the typical smell of fenugreek or curry at high concentrations and maple syrup, caramel, or burnt sugar at lower concentrations.
Several classes of MUSD have been identified that can be characterized by the percentage of normal activity of the enzyme present. In the classic type there is 0-2% normal enzymatic activity. A well-known example exists in the Mennonite community of Lancaster County, PA. in which the disease exists in 1 per 178 - 380 live births (the Hardy-Weinberg carrier frequency is 1/7 - 1/9) — the world-wide incidence is 1/185,000 live births. The Mennonite mutation is a single nucleotide substitution of an A for a T in the gene encoding the E1 α subunit of branched-chain α-keto acid dehydrogenase that changes a Tyrosine to an Asparagine.
Other classes are intermediate, with 3-30% normal enzymatic activity, intermittent, with 5-20% normal enzymatic activity, thymine-responsive, with 2-40% normal enzymatic activity, and lipoamide dehydrogenase deficiency, with 0-25% normal enzymatic activity — this is a deficiency of the E3 subunit, which also affects pyruvate dehydrogenase and α-ketoglutarate dehydrogenase because the E3 subunit is the common subunit of all three branched-chain α-Ketoacid Dehydrogenases.
The intermittent class is particularly interesting because symptoms appear under conditions of chronic stress or when the diet supplies more branched-chain amino acids than can be accommodated by the existing branched-chain α-keto acid dehydrogenase enzymatic activity. In chronic stress, cortisol signaling induces the breakdown of tissue (mainly muscle) protein, increasing the amount of available amino acids, including the branched-chain amino acids, which are metabolized predominantly in extra-hepatic tissues.