METHIONINE METABOLISM
Methionine is an important donor of a methyl group in several single-carbon transfer reactions and of the sulfur atom in the synthesis of the non-essential amino acid cysteine. After donating its methyl group, methionine is converted to homocysteine, a compound that is associated with risk of cardiovascular disease and neurological disorders. The metabolism of homocyteine requires the vitamins B6, B12, and folate.Click The Image
Methionine Is A Methyl Group Donor

In addition to its function as a substrate for protein synthesis, methionine is also an important source of a methyl group in many single-carbon transfer reactions and of the sulfur atom in the synthesis of the non-essential amino acid cysteine.
S-Adenosylmethionine (SAM)
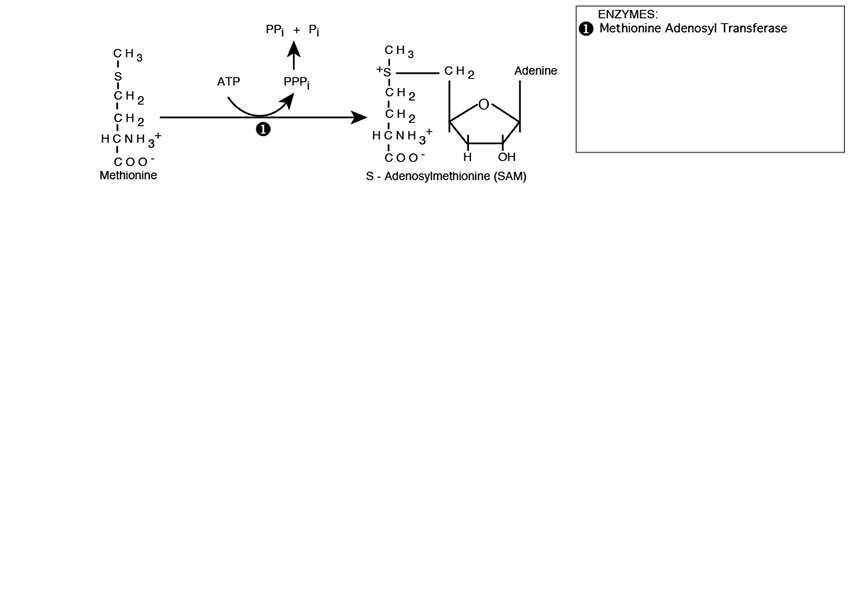
Methionine is activated for methyl donation by reacting with Adenosine Triphosphate (ATP). The methionine sulfur makes a nucleophilic attack on the ATP 5’ carbon catalyzed by Methionine Adenosyl Transferase, creating S-Adenosylmethionine (SAM) with a positively charged sulfonium center.
Methyl Transfer
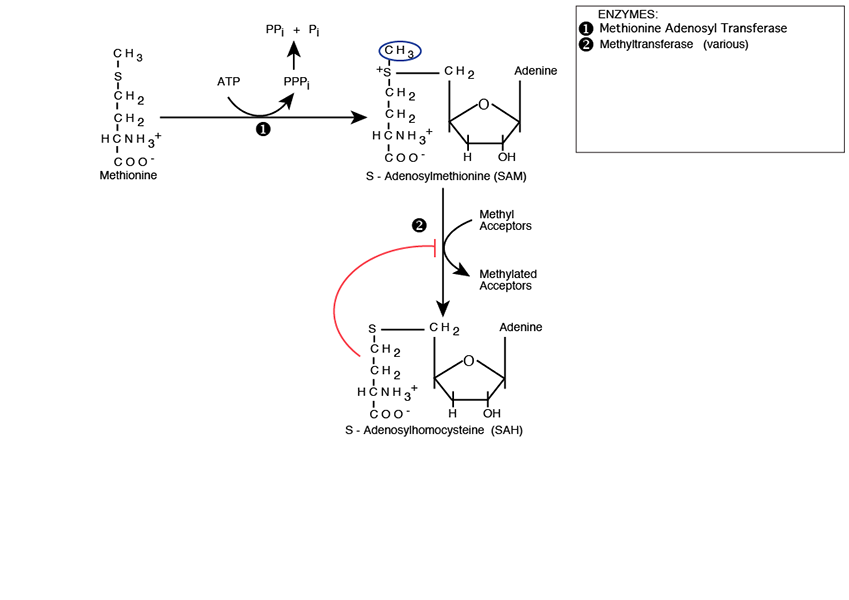
The S-Adenosylmethionine methyl group is now available for transfer by a large number of Methyltransferases, each named for its specific methyl acceptor, e.g., C-5 cytosine-specific DNA methylase, also known as DNA (cytosine-5) Methyltransferase (Dnmt1), which methylates the C-5 carbon of cytosines in DNA to produce C5-methylcytosine. The reaction product is S-Adenosylhomocyateine (SAH — SAM absent its methyl group). SAH feedback inhibits methyltransferases.
Processing Of S-Adenosylhomocysteine
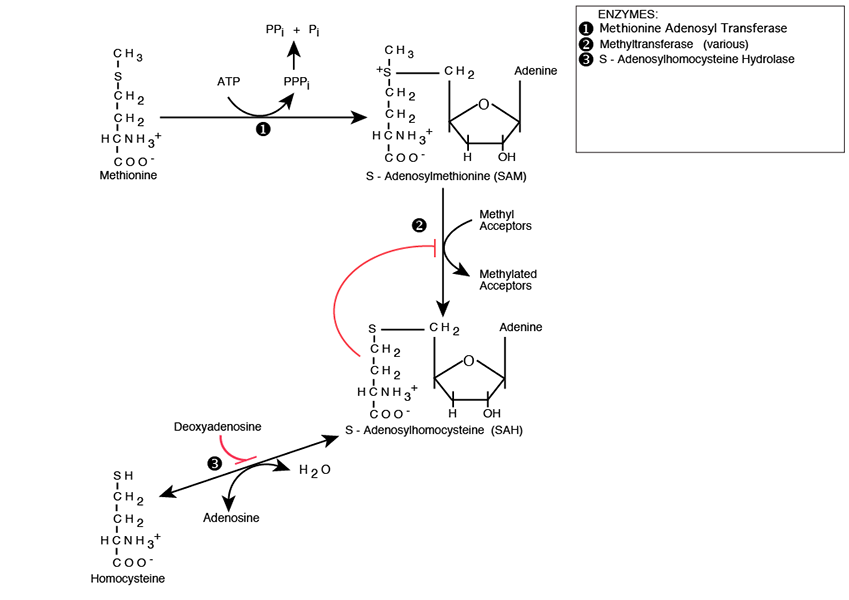
S-adenosylhomocysteine is hydrolyzed to Homocysteine and Adenosine by S-Adenosylhomocysteine Hydrolase, which is inhibited by Deoxyadenosine. Deoxyadenosine accumulation, which occurs, for example, in the autosomal recessive genetic disease Adenosine Deaminase Deficiency causes S-Adenosylhomocystein to accumulate and inhibit methyltransferases. Homocysteine can take either of two paths, Transsulfuration or Remethylation
Homocysteine On The Transsulfuration Pathway
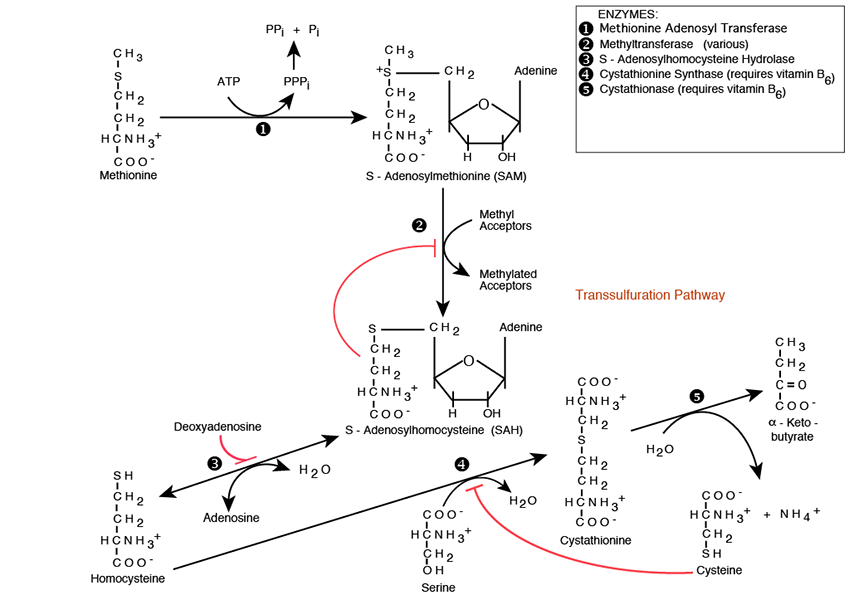
Cystathionine Synthase and Cystathionase convert Serine and Homocysteine into the non-essential amino acid Cysteine and α-Ketobutyrate; Serine, another non-essential amino acid, provides the carbon skeleton, Homocysteine provides the sulfur. Both Cystathionine Synthase and Cystathionase require vitamin B6 (Pyridoxine) as an essential co-factor. The Cystationase reaction releases NH3 -> NH4+
Metabolism of α-Ketobutyrate; Vitamin B12
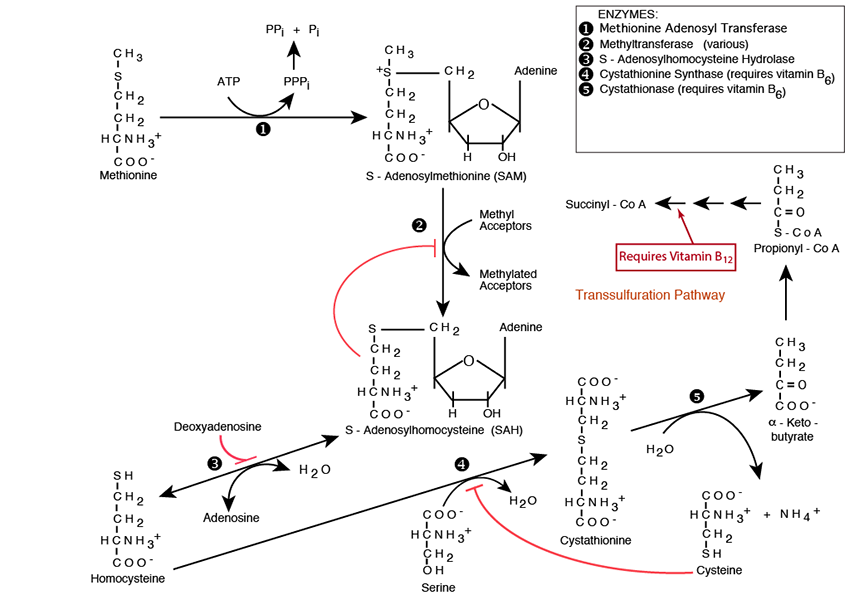
α-Ketobutyrate is converted to propionyl CoA, the same molecule as the end-product of odd-chain fatty acid β-oxidation. The propionyl CoA is converted in three enzymatic steps, one of which is one of only two reactions in Humans that required vitamin B12, to Succinyl CoA, an intermediate of the TCA cycle. More propionyl CoA is converted to succinyl CoA by this pathway from amino acid degradation than from odd-chain fatty acid β-oxidation.
Homocysteine On The Remethylation Pathway
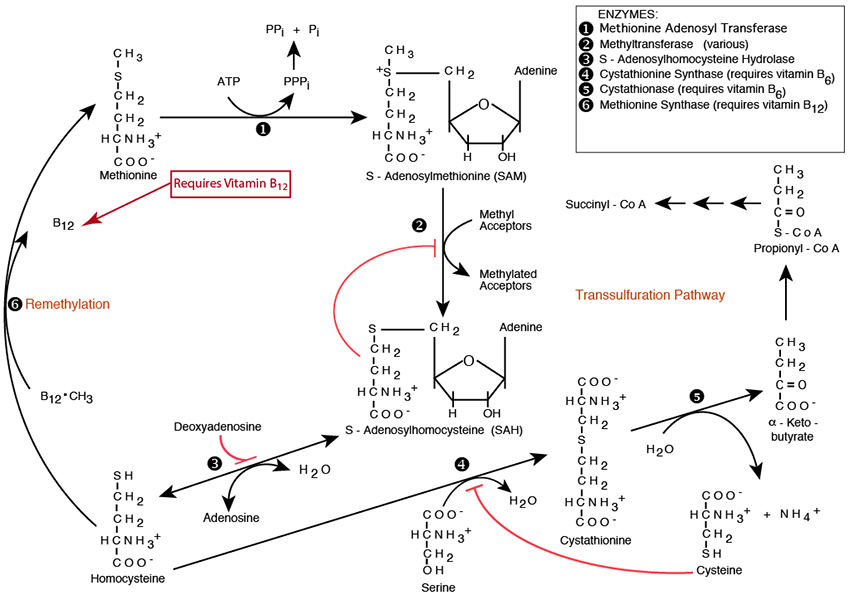
Methionine is regenerated by Methionine Synthase, which remethylates homocysteine in one of only two reactions in Humans that require vitamin B12 as the immediate methyl donor
N-5-Methyl Tetrahydrofolate Is The Primary Methyl Donor
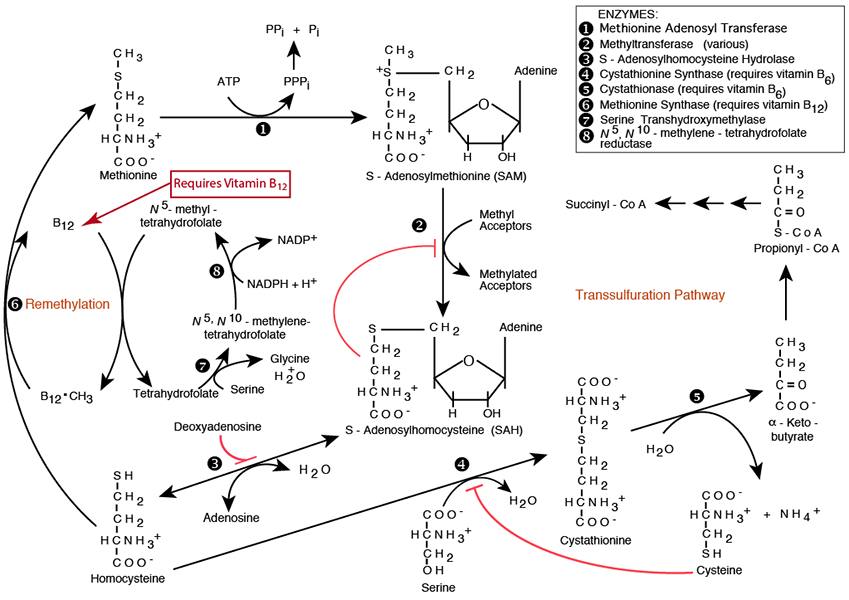
Tetrahydrofolate (see “Folate” in the top menu) is a derived from the vitamin folate (vitamin B9. It accepts and donates single carbons at the oxidation level of formaldehyde, and formic acid in many single-carbon transfer reactions. Serine is the major donor of a single carbon to tetrhydrofolate at the oxidation level of formaldehyde via Serine Transhydroxymethylase. The methylene carbon is reduced to the oxidation level of methanol, and donated to vitamin B12, which then donates it for the remethylation of Homocysteine to Methionine.
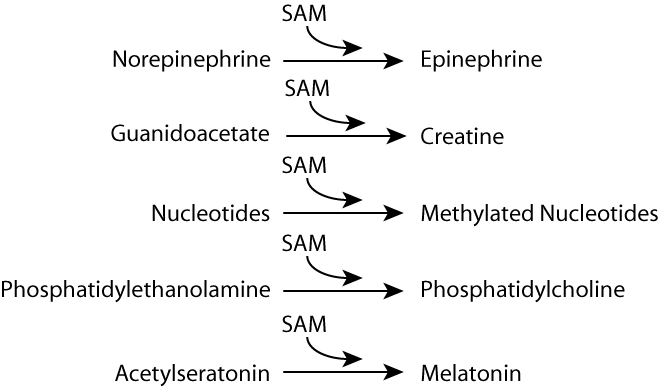
Some Specific SAM Reactions
Hyperhomocysteinemia, increased levels of homocysteine, has been shown to be associated with cardiovascular and neurologic disease, although with respect to cardiovascular disease it is controversial as to whether hyperhomocystenimia is causative. It is known that increased homocysteine can damage the endothelial lining of blood vessels. Some of the presumed mechanisms include an increase in proliferation of vascular smooth muscle cells, endothelial dysfunction, oxidative damage, an increase is synthesis of collagen and deterioration of arterial wall elastic material. Men with plasma homocysteine concentrations 12% above the upper limit of normal were determined to have approximately a threefold increase in the risk of myocardial infarction, as compared with those with lower levels. Treatment of hyperhomocysteinemia varies with the underlying cause. However, vitamin supplementation (folic acid, vitamin B6 and vitamin B12) is generally effective in reducing homocysteine concentrations. In most patients, 1 to 5 mg./day of folate rapidly decreases homocysteine concentrations. Folic acid alone, folic acid combined with vitamins B12 and B6, and vitamins B6 and B12 have all been shown to reduce homocysteine concentrations. The reduction in mortality from cardiovascular causes since 1960 has been correlated with the increase in vitamin B6 supplementation in the food supply. Genetic factors, including deficiencies in enzymes for methionine synthase, cystathionine synthase, cystathionase, enzymes involved in folate metabolism, and proteins required for folate, vitamin B6 or vitamin B12 (e.g.,intrinsic factor) absorption can all contribute to hyperhomocysteinemia, and to an increased risk of cardiovascular disease. The deficiencies of methyl tetrahydrofolate, or of methyl B12 are due either to an inadequate dietary intake of folate or B12, or to defective enzymes involved in joining methyl groups to tetrahydrofolate, transferring methyl groups from methyl tetrahydrofolate to B12, or passing them from B12 to homocysteine to form methionine.